D- STEROID HORMONES
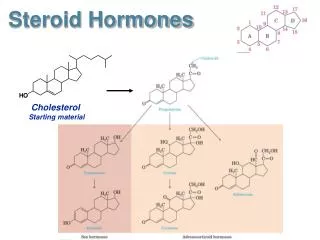
D- STEROID HORMONES. Cholesterol is the precursor of five classes of steroid hormones: glucocorticoids (for example, cortisol), mineralocorticoids (for example, aldosterone), and the sex hormones - the androgens, estrogens, and progestins.


D- STEROID HORMONES
E N D
Presentation Transcript
Cholesterol is the precursor of five classes of steroid hormones: glucocorticoids (for example, cortisol), mineralocorticoids (for example, aldosterone), and the sex hormones - the androgens, estrogens, and progestins.
Steroid hormones are transported by the blood from their sites of synthesis to their target organs. Because of their hydrophobicity, they must be complexed with a plasma protein; for example, plasma albumin can act as a nonspecific carrier for the steroid hormones. Specific plasma steroid-carrier proteins bind the steroid hormones more tightly than does albumin; for example, transcortin is responsible for transporting cortisol and corticosterone, and sex hormone-binding protein transports the sex steroids.
Synthesis of steroid hormones Synthesis involves shortening of the hydrocarbon chain of cholesterol and hydroxylation of the steroid nucleus. The initial reaction, catalyzed by the desmolase complex, converts cholesterol to pregnenolone. This is the rate-limiting step in steroid hormone biosynthesis, and requires NADPH and molecular oxygen. All the steroid hormones are derived from pregnenolone. Pregnenolone is next oxidized and then isomerized to progesterone, which is further modified by hydroxylation reactions to other steroid hormones.
Secretion of adrenal steroid hormones Adrenal cortical hormone secretion is controlled by the hypothalamus, to which the pituitary gland is attached. When the body is stressed, corticotropin-releasing factor (CRF) produced by the hypothalamus travels through a network of capillaries to the anterior lobe of the pituitary, where it induces the production and secretion of adrenocorticotropic hormone (ACTH, adrenocorticotropin). ACTH, often called the "stress hormone," stimulates the adrenal cortex to synthesize and secrete the glucocorticoid, cortisol.
Cortisol This adrenal cortical steroid binds to specific cytosolic receptors in target cells such as fibroblasts and hepatocytes. The hormone-receptor complex enters the nucleus, where it regulates specific gene expression cortisol generally stimulates the degradation of proteins to amino acids in the skeletal muscle and the promotion of gluco-neogenesis in the live.
Aldosterone Secretion of aldosterone from the adrenal cortex is induced not by ACTH but by the Na+/K+ ratio of the body and by angiotensin, an octapeptide produced in the blood from an inactive precursor (angiotensinogen) by the enzyme renin, secreted by the kidney. Aldosterone's primary effect is on the kidney tubules, where it stimulates sodium retention and potassium excretion.
Secretion of steroid hormones from gonads The testes and ovaries synthesize hormones necessary for physical development and reproduction. A single hypothalamic releasing factor, gonadotropin-releasing hormone, stimulates the anterior pituitary to release the glycoproteins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH). LH stimulates the testis to produce androgens and the ovaries to produce estrogens and progesterone. FSH regulates the growth of ovarian follicles and stimulates testicular spermatogenesis. For maximum effect on the male or female gonad, FSH also requires the presence of LH.
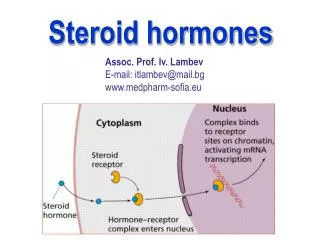
Mechanism of steroid hormone action Each steroid hormone crosses the cell membrane and binds to a specific cytosolic or nuclear receptor (hormone-dependent transcriptional factor). These receptor-hormone complexes accumulate in the nucleus and bind to regulatory DNA sequences (hormone-responsive elements, HRE), thereby causing either stimulation or inhibition of activity of specific genes. An HRE is found in each enhancer region located near a gene or group of genes that respond to a specific hormone.
Further metabolism of steroid hormones Steroid hormones are generally converted into metabolic excretion products in the liver. Reactions include reduction of unsaturated bonds and the introduction of additional hydroxyl groups. The resulting structures are made more soluble by conjugation with glucuronic acid or sulfate. Approximately 20% to 30% of these metabolites are secreted into the bile and then excreted in the feces, whereas the remainder are released into the blood and are filtered from the plasma in the kidney, passing into the urine. These conjugated metabolites are fairly water soluble and do not need protein carriers.
NITROGEN METABOLISM Amino acids contain nitrogen in addition to the carbon, hydrogen, and oxygen atoms also found in carbohydrates and fats. This nitrogen cannot be stored, and amino acids in excess of the biosynthetic needs of the cell are immediately degraded. The first phase of catabolism involves the removal of the α-amino groups by transamination and oxidative deamination, forming ammonia and the corresponding α-ketoacids. A portion of the free ammonia is excreted in the urine, but most is used in the synthesis of urea, which is quantitatively the most important route for disposing of nitrogen from the body.
In the second phase of amino acid catabolism, the carbon skeletons of the α-ketoacids are converted to common intermediates of energy-producing metabolic pathways. These compounds can be metabolized to CO2 and water, glucose, fatty acids, or ketone bodies by the central pathways of metabolism. Amino acid catabolism is part of the larger process of whole body nitrogen metabolism. Nitrogen enters the body in a variety of compounds present in the food, the most important being amino acids contained in dietary protein. Nitrogen leaves the body as urea.
Amino acids released by hydrolysis of dietary or tissue protein mix with other free amino acids distributed throughout the body, and collectively constitute the amino acid pool. Most proteins in the body are constantly being synthesized and then degraded. The total amount of protein in the body remains constant, because the rate of protein synthesis is just sufficient to replace the protein that is degraded. The rate of protein turnover varies widely for individual proteins.
Because proteins have different half-lives, it is clear that protein degradation cannot be random, but rather is influenced by some structural aspect of the protein. In contrast to carbohydrates and triacylglycerols whose major function is to provide energy, the primary role of amino acids is to serve as building blocks in biosynthetic reactions, particularly the synthesis of tissue protein. Most of the nitrogen in the diet is consumed in the form of protein, typically amounting to 70 to 100 g/day
Transport of amino acids into cells The concentration of free amino acids in the extracellular fluids is significantly lower than that within the cells of the body. This concentration gradient is maintained because active transport systems, driven by the hydrolysis of ATP, are required for movement of amino acids from the extracellular space into cells. One transport system is responsible for reabsorption in kidney tubules of the amino acids cysteine, ornithine, argirnine, and lysine. In the inherited disorder cystinuria, this carrier system is defective, resulting in the appearance of all four amino acids in the urine.
Removal of nitrogen from amino acid The first step in the catabolism of all amino acids involves the removal of the α-amino group. Once removed, this nitrogen can be incorporated into other compounds or excreted. Transamination: the funneling of amino groups to glutamate The first step in the catabolism of most amino acids is the transfer of their α-amino group to α-ketoglutarate.
The products are an α-Keto acid (derived from the original amino acid) and glutamate. α-Ketoglutarate plays a unique role in amino acid metabolism by accepting the amino groups from other amino acids, thus becoming glutamate. Glutamate produced by transamination can be oxidatively deaminated, or can be used as an amino group donor in the synthesis of nonessential amino acids. This transfer of amino groups from one carbon skeleton to another is catalyzed by a family ofenzymes called aminotransferases (formerly called transaminases). All amino acids, with the exception of lysine and threonine, participate in transamination at some point in their catabolism.
Anine aminotransferase (ALT), also called glutamate-pyruvate transaminase (GPT), is present in many tissues. The reaction is readily reversible.
Aspartate aminotransferase (AST), also called glutamate: oxaloacetate transaminase (GOT), is an exception to the rule that aminotransferases funnel amino groups to form glutamate. During amino acid catabolism, aspartate aminotransferase transfers amino groups from glutamate to oxaloacetate, forming aspartate, which is itself used as a source of nitrogen in the urea cycle.
Mechanism of action of aminotransferases. All aminotransferases require the coenzyme pyridoxal phosphate (a derivative of vitamin B6), which is covalently linked to the ε-amino group of a specific lysine residue at the active site of the enzyme. Aminotransferases act by transferring the amino group of an amino acid to the pyridoxal part of the coenzyme to generate pyridoxamine phosphate. The pyridoxamine form of the coenzyme then reacts with an α-keto acid to form an amino acid and regenerates the original aldehyde form of the coenzyme.
Diagnostic value of plasma aminotransferases Aminotransferases are normally intracellular enzymes. The presence of elevated levels of aminotransferases in the plasma indicates damage to cells rich in these enzymes. Physical trauma or a disease process can cause cell lysis, resulting in release of intracellular enzymes into the blood. Plasma AST and ALT are elevated in nearly all liver diseases, but are particularly high in conditions that cause extensive cell necrosis, such as severe viral hepatitis and prolonged circulatory collapse. Aminotransferases may be elevated in nonhepatic disease, such as myocardial infarction and muscle disorders; however, these disorders can usually be distinguished clinically from liver disease.
Oxidative deamination In contrast to transamination reactions that transfer amino groups, oxidative deamination results in the liberation of the amino group as free ammonia. These reactions occur primarily in the liver and kidney and provide α-ketoacids (which can enter the central pathway of energy metabolism) and ammonia (which is a source of nitrogen in urea synthesis). Glutamate dehydrogenase Glutamate is unique in that it is the only amino acid that undergoes rapid oxidative deamination, a reaction catalyzed by glutamate dehydrogenase.
Glutamate dehydrogenase is unusual in that it can use either NAD+ or NADP+ as a coenzyme. ATP and GTP are allosteric inhibitors of glutamate dehydrogenase, whereas GDP and ADP are activators of the enzyme.
UREA CYCLE Urea is the major disposal form of amino groups derived from amino acids, and accounts for about 90% of the nitrogen-containing components of urine. One nitrogen of the urea molecule is supplied by free NH3 and the other nitrogen by aspartate. Glutamate is the immediate precursor of both ammonia (through oxidative deamination by glutamate dehydrogenase) and aspartate nitrogen (through transamination of oxaloacetate by aspartate aminotransferase). The carbon and oxygen of urea are derived from CO2. Urea is produced by the liver and then is transported in the blood to the kidneys for excretion in the urine. The first two reactions leading to the synthesis of urea occur in the mitochondria, whereas the remaining cycle enzymes are located in the cytosol. Glutamate dehydrogenase also occurs in the mitochondria, providing ammonia for incorporation into carbamoyl phosphate.
Urea diffuses from the liver and is transported in the blood to the kidneys, where it is filtered and excreted in the urine. A portion of the urea synthesized in the liver diffuses from the blood into the intestine and is cleaved to CO2 and NH3 by bacterial urease. This ammonia is partly lost in the feces and is partly reabsorbed into the blood. In patients with kidney failure, plasma urea levels are elevated, promoting a greater transfer of urea from blood into the gut. The intestinal action of urease on this urea becomes a clinically important source of ammonia, contributing to the hyperammonemia often seen in these patients.
Regulation of the urea cycle N-Acetylglutamate is an essential activator for carbamoyl phosphate synthetase I, the rate-limiting step in the urea cycle. METABOLISM OF AMMONIA Although ammonia is involved in the formation of urea in the liver, the level of ammonia in the blood must be kept low because even slightly elevated concentrations (hyperammonemia) are toxic to the central nervous system. From amino acids Many tissues, but particularly the liver, form ammonia from amino acids by the aminotransferase and glutamate dehydrogenase reactions.
From glutamine The kidneys form ammonia from glutamine by the action of renal glutaminase. Most of this ammonia is excreted into the urine as NH4+, which is an important mechanism for maintaining the body's acid-base balance. Ammonia is also obtained from the hydrolysis of glutamine by. From bacterial action in the intestine: Ammonia is formed by the bacterial degradation of urea in the lumen of the intestine. Ammonia is absorbed from the intestine by way of the portal vein and is almost quantitatively removed by the liver by conversion to urea. From amines: Amines obtained from the diet and monoamines that serve as hormones or neurotransmitters give rise to ammonia by the action of amine oxidase.
From purines and pyrimidines: In the catabolism of purines and pyrimidines, amino groups attached to the rings are released as ammonia. Although ammonia is constantly produced in the tissues, it is present at very low levels in blood. This is due to both the rapid removal of ammonia from the blood by the liver and the fact that many tissues, particularly muscle, release amino acid nitrogen in the form of glutamine or alanine, rather than as free ammonia. Formation of urea in the liver is quantitatively the most important disposal route for ammonia. This amide of glutamic acid provides a nontoxic storage and transport form of ammonia. The formation of glutamine occurs primarily in the muscle and liver but also is important in the nervous system, where it is the major mechanism for the removal of ammonia in the brain.
Elevated concentrations of ammonia in the blood cause the symptoms of ammonia intoxication, which include tremors, slurring of speech, and blurring of vision. Cirrhosis of the liver caused by alcoholism, hepatitis, or biliary obstruction may result in formation of collateral circulation around the liver. Genetic deficiencies of each of the five enzymes of the urea cycle. Failure to synthesize urea leads to hyperammonemia during the first week following birth. All inherited deficiencies of the urea cycle enzymes result in mental retardation. The toxicity of high levels of ammonia is thought to result, in part, from a shift in the equilibrium of the glutamate dehydrogenase reaction toward the direction of glutamate formation:
This depletes α-ketoglutarate, an essential intermediate in the citric acid cycle, resulting in a decrease in cellular oxidation and ATP production. The brain is particularly vulnerable to hyperammonemia, presumably because it depends on the citric acid cycle to maintain its high rate of energy production.
This depletes α-ketoglutarate, an essential intermediate in the citric acid cycle, resulting in a decrease in cellular oxidation and ATP production. The brain is particularly vulnerable to hyperammonemia, presumably because it depends on the citric acid cycle to maintain its high rate of energy production.