Download

1 / 26
260 likes | 363 Vues
Explore the impact of hotspots and cold spots on chromosome segregation, gene evolution, and other DNA-related processes in yeast meiosis. Investigate conservation, essential genes, base usage, motifs, and more.

E N D
Alessandro Brozzi 19 November 2007 Wolfgang Huber Group -Internal Meeting- Association of event rates with genome properties
Introduction In the previous sections of the current project about the mapping of all the recombination events that occurred in yeast meiosis, the methodologies and the analysis to identify regions of high (hot spots) and low (cold spots) recombination have been described. Different sets of these region are now available either for the wt and mutant strain. The mechanism of initiating recombination cannot however be understood without knowledge of the factors and the combination of molecular features that regulate hot spots and cold spots. The understanding of the hot spots and cold spots will be relevant to comprehending in details the molecular processes leading to accurate chromosome segregation, assembling new configurations of physically linked genes during evolution and generally to comprehending other DNA-related processes that are affected by chromosome context, such as transcription and replication.
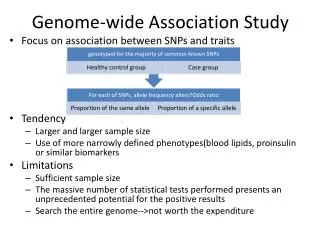
Outline Genomic and genetic properties subjects of investigation: • Interspecies conservation • Essential genes • Base usage • Motifs • Ty element • Deletions • Go categories • (nucleosome positioning)
Interspecies conservation To evaluate whether or not hotspots were located in conserved genomic regions, I looked at the interpsecies conservation. Raw data "Conservation" track from UCSC which represents a measure of evolutionary conservation in seven species of the genus Saccharomyces based on a phylogenetic hidden Markov model(phastCons). These are the species: * S. paradoxus * S. mikatae * S. kudriavzevii * S. bayanus * S. castelli * S. kluyveri It ranges from 1: high conservation to 0: poor conservation
Interspecies conservation Raw data • Complex events 2.5 fixed Processing • Identification of hot spots Datasets • Total recombination events hot spots defined by a p-value cutoff of 10^-3 • X hot spots defined by a p-value cutoff of 10^-3 • C hot spots defined by a p-value cutoff of 10^-3 Analysis • Calculate a genome wide distribution of the conservation scores • Match each hot spot interval with the conservation scores • Match different sets of flanking regionswith the conservation scores 250 bps hot spot Downstream flanking regions Upstream flanking regions
Interspecies conservation see more
Essential genes • Essential Genes (18% of the 6200 S. cerevisiae genes) are the genes that are indispensable to sustain cellular life. The functions encoded by essential genes are considered as a foundation of life and therefore are likely to be common for all cells. Raw data • A list of essential genes provided me by Eugenio • Genome Features Annotation table download by SGD Processing • Conversion to alignment system • Match with the list and annotation of each ORFs (6346) as essential and non essential • 1169 resulting essential genes Analysis • Mapping of the essential genes below the rate events plot • Measure of the association with the hot psots
Essential genes - plots and p-values - see more [1] "total_p_3_ext0" Overalpping Not Overlapping Essential 36 1105 Not Essential 238 4967 file p.value X_p_3_ext0 0.06169657 <-- X_p_3_ext5000 0.18158298 X_p_3_ext10000 0.86689951 C_p_3_ext0 0.90457358 C_p_3_ext5000 0.95425873 C_p_3_ext1000 0.64005255 total_p_3_ext0 0.03613371 <-- total_p_3_ext5000 0.72022013 total_p_3_ext1000 0.09765762 <-- two sided Fisher test
Base usage - intro - It is possible that due to different mismatch repair mechanisms directional biases in base composition in the hot spot included genomic regions can be produced. Raw data • Complex annotation fixed data 2.5 • Alignment summary Processing • Identification of hot spots • Conversion to S288c coordinates to retrieve the sequences Datasets • Total recombination events hot spots defined by a p-value cutoff of 10^-3 • X hot spots defined by a p-value cutoff of 10^-3 • C hot spots defined by a p-value cutoff of 10^-3 Analysis • Compute the base compositions in hot spots regions • Compare with the null expected base usage distributions
Base usage (not combined) see more
Base usage (combined ac/gt) see more
Base usage (p-values) Not combined (a,c,g,t) > S288c_global=c(0.310,0.191,0.191,0.308) > chisq.test( total_p_3_base_usage, p = S288c_global) Chi-squared test for given probabilities data: total_p_3_base_usage X-squared = 17.6811, df = 3, p-value = 0.0005117 X-squared df p-value X_p_3_base_usage 17.5454 3 0.0005458 C_p_3_base_usage 9.8913 3 0.01951 Combined pairs at/gc p_null_combined=c(0.5123,0.4877) X-squared df p-value total_p_3_base_usage_combined 24.0833 1 9.226e-07 X_p_3_base_usage_combined 24.7047 1 6.682e-07 C_p_3_base_usage_combined 17.8944 1 2.335e-05
Motifs To find putative sequence motifs characterizing the hostspots, I submitted to MEME each dataset of DNA sequences retrieved from the central region of each hotspot (from 100 to 300 bps). I used the following parameters: • 3 as maximum number of motifs to find in both strands with any number of repetitions per sequence • minumum motif width 6 bps and maximum motif width 20 bps • stop if motif E-value greater 1e100. • background model: 0-order Markov model based on the letter frequencies in the training set. Raw data • Complex annotation fixed data 2.5 Datasets • Total recombination events hot spots defined by a p-value cutoff of 10^-3 • X hot spots defined by a p-value cutoff of 10^-3 • C hot spots defined by a p-value cutoff of 10^-3 see more
Ty elements Ty elements are 6 kb long retrotransposons flanked by two long terminal repeats (LTRs). Tys are present in 30-40 copies per haploid cell, representing 3% of the genome They are structurally and functionally similar to retroviruses and have a similar life cycle Raw data • Genome Features Annotation table download by SGD • Complex annotation fixed data 2.5 Processing • Parsing Ty element information from the main table • Coordinate conversion Analysis • Mapping of the Ty elements below the rate events plot • Measure of the association with the hot spots; compare the distribution of the p-values of the intermarkers intervals at the borders of Ty element with the overall distribution of the p-values.
Deletions Raw data • Object “Polymorphisms” • Complex annotation fixed data 2.5 Processing • Fusing together small DELs into single larger DELs (at most spaced by 50 bps) • Identification of hot spots Analysis • Visualization by IGB • Measure of the association with the hot spots; compare the distribution of the p-values of the intermarkers intervals at the borders of each DELs with the overall distribution of the p-values.
GO categories Raw data • Complex annotation fixed data 2.5 • Genome Features Annotation table download by SGD Processing • GO stats package • Identification of hot spots • Parsing of the ORFs from the annotation table • Coordinated conversion in alignment coordinates Dataset • Total recombination events hot spots defined by a p-value cutoff of 10^-3 • X hot spots defined by a p-value cutoff of 10^-3 • C hot spots defined by a p-value cutoff of 10^-3 Analysis • Hyper Geometric Test to look at over represented categories of class Biological Process
Nucleosomes Nucleosomes are the structures which DNA filament is wrapped to. Raw data • Raw data of the HHM values Processing • Identification of large depletions Analysis • Mapping of the nucleosome positioning below the rate events plot • Visualization of the track by IGB
Conclusions Summary Hot spots seem to lie in rather conserved isolated genomic regions ORFs which overlap a hot spot tend to have fewer essential members than expected (marginally significant p-values) AT down representation in the base composition for the regions included in hot spots Ty elements and INDELS are associated with cold spots Lipid metabolic processes are the “biological process” categories which seem to be more enriched associated with hot spots regions