DNA Sequencing
DNA Sequencing. DNA sequencing. How we obtain the sequence of nucleotides of a species. …ACGTGACTGAGGACCGTG CGACTGAGACTGACTGGGT CTAGCTAGACTACGTTTTA TATATATATACGTCGTCGT ACTGATGACTAGATTACAG ACTGATTTAGATACCTGAC TGATTTTAAAAAAATATT…. Which representative of the species?. Which human?

DNA Sequencing
E N D
Presentation Transcript
DNA sequencing How we obtain the sequence of nucleotides of a species …ACGTGACTGAGGACCGTG CGACTGAGACTGACTGGGT CTAGCTAGACTACGTTTTA TATATATATACGTCGTCGT ACTGATGACTAGATTACAG ACTGATTTAGATACCTGAC TGATTTTAAAAAAATATT…
Which representative of the species? Which human? Answer one: Answer two: it doesn’t matter Polymorphism rate: number of letter changes between two different members of a species Humans: ~1/1,000 Other organisms have much higher polymorphism rates • Population size!
Why humans are so similar Out of Africa N A small population that interbred reduced the genetic variation Out of Africa ~ 40,000 years ago Heterozygosity: H H = 4Nu/(1 + 4Nu) u ~ 10-8, N ~ 104 H ~ 410-4
Human population migrations • Out of Africa, Replacement • “Grandma” of all humans (Eve) ~150,000yr • Ancestor of all mtDNA • “Grandpa” of all humans (Adam) ~100,000yr • Ancestor of all Y-chromosomes • Multiregional Evolution • Fossil records show a continuous change of morphological features • Proponents of the theory doubt mtDNA and other genetic evidence
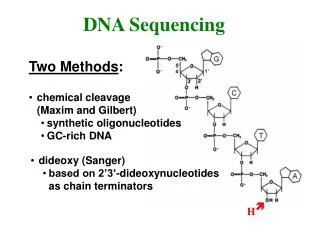
DNA Sequencing – Overview 1975 • Gel electrophoresis • Predominant, old technology by F. Sanger • Whole genome strategies • Physical mapping • Walking • Shotgun sequencing • Computational fragment assembly • The future—new sequencing technologies • Pyrosequencing, single molecule methods, … • Assembly techniques • Future variants of sequencing • Resequencing of humans • Microbial and environmental sequencing • Cancer genome sequencing 2015
DNA Sequencing Goal: Find the complete sequence of A, C, G, T’s in DNA Challenge: There is no machine that takes long DNA as an input, and gives the complete sequence as output Can only sequence ~800 letters at a time
DNA Sequencing – vectors DNA Shake DNA fragments Known location (restriction site) Vector Circular genome (bacterium, plasmid) + =
DNA Sequencing – gel electrophoresis • Start at primer (restriction site) • Grow DNA chain • Include dideoxynucleoside (modified a, c, g, t) • Stops reaction at all possible points • Separate products with length, using gel electrophoresis
Reading an electropherogram • Filtering • Smoothening • Correction for length compressions • A method for calling the letters – PHRED PHRED – PHil’s Read EDitor (by Phil Green) Newer methods may be better, but labs are reluctant to change
Output of PHRED: a read A read: 500-1000 nucleotides A C G A A T C A G …A 16 18 21 23 25 15 28 30 32 …21 Quality scores: -10log10Prob(Error) Reads can be obtained from leftmost, rightmost ends of the insert Double-barreled sequencing: (1990) Both leftmost & rightmost ends are sequenced, reads are paired
Method to sequence longer regions genomic segment cut many times at random (Shotgun) Get one or two reads from each segment ~800 bp ~800 bp
Reconstructing the Sequence (Fragment Assembly) reads Cover region with high redundancy Overlap & extend reads to reconstruct the original genomic region
Definition of Coverage C Length of genomic segment: L Number of reads: n Length of each read: l Definition:Coverage C = n l / L How much coverage is enough? Lander-Waterman model: Assuming uniform distribution of reads, C=10 results in 1 gapped region /1,000,000 nucleotides
Repeats Bacterial genomes: 5% Mammals: 50% Repeat types: • Low-Complexity DNA (e.g. ATATATATACATA…) • Microsatellite repeats (a1…ak)N where k ~ 3-6 (e.g. CAGCAGTAGCAGCACCAG) • Transposons • SINE(Short Interspersed Nuclear Elements) e.g., ALU: ~300-long, 106 copies • LINE(Long Interspersed Nuclear Elements) ~4000-long, 200,000 copies • LTRretroposons(Long Terminal Repeats (~700 bp) at each end) cousins of HIV • Gene Families genes duplicate & then diverge (paralogs) • Recent duplications ~100,000-long, very similar copies
AGTAGCACAGACTACGACGAGACGATCGTGCGAGCGACGGCGTAGTGTGCTGTACTGTCGTGTGTGTGTACTCTCCTAGTAGCACAGACTACGACGAGACGATCGTGCGAGCGACGGCGTAGTGTGCTGTACTGTCGTGTGTGTGTACTCTCCT Sequencing and Fragment Assembly 3x109 nucleotides 50% of human DNA is composed of repeats Error! Glued together two distant regions
What can we do about repeats? Two main approaches: • Cluster the reads • Link the reads
What can we do about repeats? Two main approaches: • Cluster the reads • Link the reads
What can we do about repeats? Two main approaches: • Cluster the reads • Link the reads
AGTAGCACAGACTACGACGAGACGATCGTGCGAGCGACGGCGTAGTGTGCTGTACTGTCGTGTGTGTGTACTCTCCTAGTAGCACAGACTACGACGAGACGATCGTGCGAGCGACGGCGTAGTGTGCTGTACTGTCGTGTGTGTGTACTCTCCT A R B D R C Sequencing and Fragment Assembly 3x109 nucleotides ARB, CRD or ARD, CRB ?
AGTAGCACAGACTACGACGAGACGATCGTGCGAGCGACGGCGTAGTGTGCTGTACTGTCGTGTGTGTGTACTCTCCTAGTAGCACAGACTACGACGAGACGATCGTGCGAGCGACGGCGTAGTGTGCTGTACTGTCGTGTGTGTGTACTCTCCT Sequencing and Fragment Assembly 3x109 nucleotides
Strategies for whole-genome sequencing • Hierarchical – Clone-by-clone • Break genome into many long pieces • Map each long piece onto the genome • Sequence each piece with shotgun Example: Yeast, Worm, Human, Rat • Online version of (1) – Walking • Break genome into many long pieces • Start sequencing each piece with shotgun • Construct map as you go Example: Rice genome • Whole genome shotgun One large shotgun pass on the whole genome Example: Drosophila, Human (Celera), Neurospora, Mouse, Rat, Dog
a BAC clone map Hierarchical Sequencing Strategy • Obtain a large collection of BAC clones • Map them onto the genome (Physical Mapping) • Select a minimum tiling path • Sequence each clone in the path with shotgun • Assemble • Put everything together genome
Methods of physical mapping Goal: Make a map of the locations of each clone relative to one another Use the map to select a minimal set of clones to sequence Methods: • Hybridization • Digestion
1. Hybridization Short words, the probes, attach to complementary words • Construct many probes • Treat each BAC with all probes • Record which ones attach to it • Same words attaching to BACS X, Y overlap p1 pn
2. Digestion Restriction enzymes cut DNA where specific words appear • Cut each clone separately with an enzyme • Run fragments on a gel and measure length • Clones Ca, Cb have fragments of length { li, lj, lk } overlap Double digestion: Cut with enzyme A, enzyme B, then enzymes A + B
The Walking Method • Build a very redundant library of BACs with sequenced clone-ends (cheap to build) • Sequence some “seed” clones • “Walk” from seeds using clone-ends to pick library clones that extend left & right
Walking off a Single Seed • Low redundant sequencing • Many sequential steps
Walking off a single clone is impractical • Cycle time to process one clone: 1-2 months • Grow clone • Prepare & Shear DNA • Prepare shotgun library & perform shotgun • Assemble in a computer • Close remaining gaps • A mammalian genome would need 15,000 walking steps !
Walking off several seeds in parallel • Few sequential steps • Additional redundant sequencing In general, can sequence a genome in ~5 walking steps, with <20% redundant sequencing Efficient Inefficient
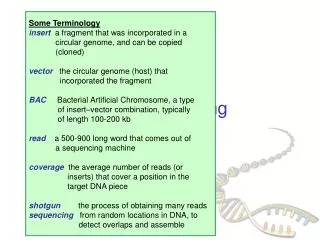
Some Terminology insert a fragment that was incorporated in a circular genome, and can be copied (cloned) vector the circular genome (host) that incorporated the fragment BACBacterial Artificial Chromosome, a type of insert–vector combination, typically of length 100-200 kb read a 500-900 long word that comes out of a sequencing machine coveragethe average number of reads (or inserts) that cover a position in the target DNA piece shotgun the process of obtaining many reads sequencing from random locations in DNA, to detect overlaps and assemble
cut many times at random Whole Genome Shotgun Sequencing genome plasmids (2 – 10 Kbp) forward-reverse paired reads known dist cosmids (40 Kbp) ~800 bp ~800 bp
Fragment Assembly Given N reads… Where N ~ 30 million… We need to use a linear-time algorithm
Steps to Assemble a Genome Some Terminology read a 500-900 long word that comes out of sequencer mate pair a pair of reads from two ends of the same insert fragment contig a contiguous sequence formed by several overlapping reads with no gaps supercontig an ordered and oriented set (scaffold) of contigs, usually by mate pairs consensus sequence derived from the sequene multiple alignment of reads in a contig 1. Find overlapping reads 2. Merge some “good” pairs of reads into longer contigs 3. Link contigs to form supercontigs 4. Derive consensus sequence ..ACGATTACAATAGGTT..
1. Find Overlapping Reads (read, pos., word, orient.) aaactgcag aactgcagt actgcagta … gtacggatc tacggatct gggcccaaa ggcccaaac gcccaaact … actgcagta ctgcagtac gtacggatc tacggatct acggatcta … ctactacac tactacaca (word, read, orient., pos.) aaactgcag aactgcagt acggatcta actgcagta actgcagta cccaaactg cggatctac ctactacac ctgcagtac ctgcagtac gcccaaact ggcccaaac gggcccaaa gtacggatc gtacggatc tacggatct tacggatct tactacaca aaactgcagtacggatct aaactgcag aactgcagt … gtacggatct tacggatct gggcccaaactgcagtac gggcccaaa ggcccaaac … actgcagta ctgcagtac gtacggatctactacaca gtacggatc tacggatct … ctactacac tactacaca