Download

1 / 20
260 likes | 1.35k Vues
Guidance for Industry CGMP for Phase 1 Investigational Drugs. July 2008 Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory Affairs (ORA). Introduction. Panel Members: Melissa Morandi, Moderator Zhengtian “Titan” Gu, PhD

E N D
Guidance for IndustryCGMP for Phase 1 Investigational Drugs July 2008 Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory Affairs (ORA)
Introduction • Panel Members: • Melissa Morandi, Moderator • Zhengtian “Titan” Gu, PhD • Lizzie Leininger , PhD • William (Rick) Srigley • Interactive Panel Format • Hold questions until last speaker • Share information about your interpretation and how your company is implementing (or not) the guidance • How will this guidance change Phase 1 manufacturing? • Will it foster more opportunities to get into Phase 1?
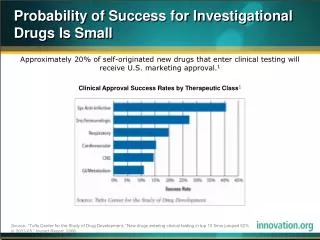
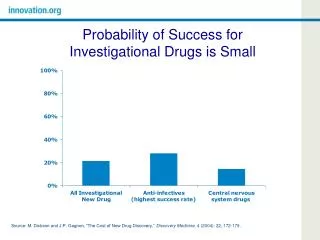
Overview: cGMP for Phase 1 • Past: Applied same standard to all drugs that were be administered to humans • July 2008: The manufacture of most investigational new drugs (IND) used in phase 1 clinical trials are exempt from complying with 21 CFR part 211 • Improve the quality of phase 1 investigational drugs and facilitate the initiate of clinical trials in humans
Overview: cGMP for Phase 1 • Other guidance from FDA: • “INDs---Approaches to Complying with CGMP During Phase 1” dated January 2006 is being issued concurrently with a final rule that specifies that 21 CFR part 211 no longer applies for most investigational products, including exploratory products that are manufactured for use in phase 1 • Preamble to 21 CFR 210 and 211 • 505(i) of the FD&C Act
Overview: cGMP for Phase 1 • Included: • Recombinant and non-recombinant products • Vaccines • Allergenic products • In vivo diagnostics • Plasma derivatives Blood and blood components (but must comply with 21 CFR 600-660) • Gene therapy and somatic cellular therapy • APIs used in phase 1 • Excluded: • Human cell or tissue products • Clinical trials for device approval • Products manufactured for phase 1 and 2 clinical trials • Already approved products • PET drugs (21 CFR 212)
Overview: CGMP for Phase 1 • General Points throughout the guideline: • Quality and safety maintained by appropriate QC procedures, Good scientific and QC principles • Well-defined, written procedures • Adequately controlled equipment and manufacturing environment • Accurate and consistently recorded data from manufacturing and testing • Drugs must meet appropriate standards of safety, identity, strength, quality, and purity • Use of the word appropriate ~ 35 times
Facilities • Can use laboratories that are not expressly or solely designed for their manufacture, can use shared facilities, academic institutions • Need to conduct a comprehensive and systematic evaluation of the manufacturing setting to identify potential hazards • Focus on contamination and cross-contamination with other substances • Identify appropriate actions prior to and during manufacturing that eliminate or mitigate those hazards • Assess contract facilities to ensure effective quality control functions • How will we document these assessments?
Equipment • Equipment: Disposables, Prepackaged Materials, Closed Systems (can potentially alleviate the need for stricter room classification…) • “Appropriate” space, lighting, cooling, ventilation, cooling, heating, plumbing, washing, and sanitation • Identify all equipment used for a particular process and document it in the record • Equipment that will not react with, add to, or be absorbed by the phase 1 drug • Equipment properly maintained, cleaned, calibrated and sanitized following written procedures • Procedural controls to prevent contamination and cross contamination • Air cleanliness suitable to the operations “Use of procedural controls in a facility promotes orderly manufacturing and aids in preventing contamination…”
Personnel and the QC Function • Personnel need education, experience and training • Need a written plan that describes the role and responsibilities of the QC function • Examine materials and components • Review and approval of manufacturing and testing procedures and acceptance criteria • Releasing or rejecting batches • Investigating unexpected results or errors • Recommends assigning an individual independent of manufacturing, however…admits that “ in some small operations it may be necessary to have the same individual perform the manufacturing and the QC functions” • Another qualified individual independent of manufacturing must conduct periodic reviews
Materials • Traceable to each batch • Records need: receipt date, quantity of shipment. supplier’s name, lot number, storage conditions, and expiration date • Establish criteria and examine C of A • Identity testing for API (does not mention identity testing as a requirement for other materials)
Documentation • General Records: • Equipment maintenance and calibration • Manufacturing records and analytical test records • Distribution records • QC Function Plan • Component records • Deviations and investigations • Complaints
Documentation • Sufficient records to replicate manufacturing: • Materials • Equipment • Procedures used • Any problems encountered in manufacturing • Record of changes • Record of microbial controls
Laboratory Controls • Scientifically sound: specific, sensitive and accurate (but does not use the word qualification…) • Specifications for known safety-related concerns should be established and met • Personnel verify that equipment is in good working condition (e.g. system suitability) • Stability from date of manufacture through date of last administration
Packaging, Labeling and Distribution • Procedures for product segregation, label reconciliation, verification of operations by a second person, confirmatory laboratory testing and QC review • Distribution record sufficiently detailed to allow traceability and facilitate recall.
Special Manufacturing Situations • Multi-Product Facilities: Periodically evaluate the implemented procedural control for their effectiveness • Biologics: • Testing to reproduce a comparable phase 1 drug, in-process and final product retains for comparison • Equipment and controls for safety-related functions (e.g. viral clearance) • Testing for safety-related purposes (viral loads, bioburden, removal of residual substances, etc.) • Adventitious agent control • Alternate approaches allowed for gene therapy (e.g. one batch per patient)
Aseptic Processing • References FDA “Guidance for Industry: Sterile Drugs Produced by Aseptic Processing-Current Good Manufacturing Practices.”
cGMPs for Phase 1 Other Considerations • Where is the clinical trial? • ICH Q7A • EMEA • Raw material identity testing • Quality Agreements • Quality Unit • Facility • QP Audits, release of lots, and stability extensions • Expectations of other territories • Expectations of potential partners • What else will the material be used for? • Is it a phase 1/phase 2 trial
cGMPs for Phase 1 Other Considerations • Drug device combination products • Need to follow QSR and Design Controls • Quality Systems and documentation: • Use same or different Quality System for Phase 1 • Separate Training for Phase 1 • Assay Qualification • Audit Program • Document Review • Staffing • Additional requirements such as QC plan, hazard assessment, multi-product use periodic assessment of procedural control, • Documentation of scientific rational • Where will phase 2 work be done? Is there enough information to ensure an efficient and successful tech transfer?
Contract Manufacturing and Testing • Different Quality Agreement for phase 1contractors, or Development Agreement or Scope of Work? • Can you use the pilot or non-GMP facility at contractors? (less $$$ and time constraints) • Documentation practices at contractor • Deviation and Change approval practices • Assessment or Audit • Use sites you may have not used previously • New facilities specific to Phase 1
Impact to Timeline? • Flexibility in facilities uses for manufacturing, internal and for contract manufacturing • Flexibility in staffing • Disposable equipment, less cleaning validation and change over time for facilities • Assay qualification • Documentation in the guideline is similar, but more specific in some areas than 21CFR 211