ChIP-Seq
ChIP-Seq. Giulio Pavesi University of Milano giulio.pavesi@unimi.it. Epigenetics. Modern experimental techniques and technologies allow for the genome-wide study of different types of histone modifications, shedding light on the role of each one. “ChIP”.

ChIP-Seq
E N D
Presentation Transcript
ChIP-Seq Giulio Pavesi University of Milano giulio.pavesi@unimi.it
Epigenetics • Modern experimental techniques and technologies allow for the genome-wide study of different types of histone modifications, shedding light on the role of each one
“ChIP” • If we have the “right” antibody, we can extract (“immunoprecipitate”) from living cells the protein of interest bound to the DNA • And - we can try to identify which were the DNA regions bound by the protein • Can be done for transcription factors • But can be done also for histones - and separately for each modification
After ChIP Identification of the DNA fragment bound by the protein PCR Microarray Hybridization Sequencing
Many cells- many copies of the same region bound by the protein
ChIP-Seq Histone ChIP TF ChIP
So - if we found that a region has been sequenced many times, then we can suppose that it was bound by the protein, but…
Only a short fragment of the extracted DNA region can be sequenced, at either or both ends (“single” vs “paired end” sequencing) for no more than 35 (before) / 50 (now) / 75 (now) bps Thus, original regions have to be “reconstructed” …and, once again, bioinformaticians can be of help…
“Peak finding”, in a perfect world Peaks: How tall? How wide? How much enriched?
“Peak finding” • The main issue: the DNA sample sequenced (apart from sequencing errors/artifacts) contains a lot of “noise” • Sample “contamination” - the DNA of the PhD student performing the experiment • DNA shearing is not uniform: open chromatin regions tend to be fragmented more easily and thus are more likely to be sequenced • Repetitive sequences might be artificially enriched due to inaccuracies in genome assembly • Amplification pushed too much: you see a single DNA fragment amplified, not enriched • As yet unknown problems, that anyway seem to produce “noisy” sequencings and screw the experiment up
Dealing with “noise” • A solution is to perform a “control” experiment, and use it to filter out the results: • “Input DNA”: a portion of the DNA sample removed before IP • “Mock IP DNA”: DNA obtained from IP without antibodies • “Non-specific IP DNA”: DNA obtained from IP using an antibody against a protein not known to be involved in DNA binding, e.g. immunoglobulin G • “Knock out IP DNA”: perform IP as in the main experiment, but on “knock out” cells that do not express the TF studied (cannot be done for histones…)
12 different methods have been introduced in less than 2 years - each one answers the questions in a different way and computes significance (p or q values) with different strategies They output the list of “enriched regions” from your experiment
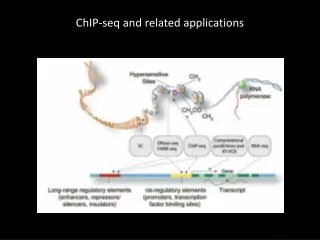
Histone modifications at transcribed regions Read count (peak height) High Low Expression level
Insulator regions Enhancer regions
Not only methylations… • Wang et. al, Nature Genetics 40(7), 2008 • 18 histone acetylations • H2AK5ac • H2BK5ac • H2BK12ac • H2BK20ac • H2BK120ac • H3K4ac • H3K9ac • H3K14ac • H3K18ac • H3K23ac • H3K27ac…. • And intersection with methylation maps
“patterns” of modifications associated with promoters of genes
Histones in different cell types • Mikkelsen et. al., Nature 448, 553-560, 2007 • Chip-Seq in three mouse cell types: • Embryonic Stem (ES) cells • Neural Progenitor Cells (NPCs) • Embryonic Fibroblasts (MEFs) • Produced over 4G sequences • For histone modifications: • H3K4me3, H3K9me3, H3K27me3, H3K36me3, H4K20me3 • And • RNA polymerase II
Neural TF Housekeeping Neurogenesis TF Adipogenesis TF NP Marker gene H3K4me3+H3K27me3 is a “bivalent” chromatin mark, Poises genes for lineage specific activation or repression
Histone Data at the UCSC • The ENCODE project is producing whole-genome maps of • Transcription factors • DNA methylation • CTCF/insulators • PolII/III binding • Histone modifications • ..across several different cell lines • As data are produced, they are loaded as “tracks” accessible and retrievable through the UCSC genome browser
What next? • From the signature histone modifications, “characterize” genomic regions: • Promoter? • Enhancer? • Transcribed? • Repressed?
Cell-type-specific promoter and enhancer states and associated functional enrichments.