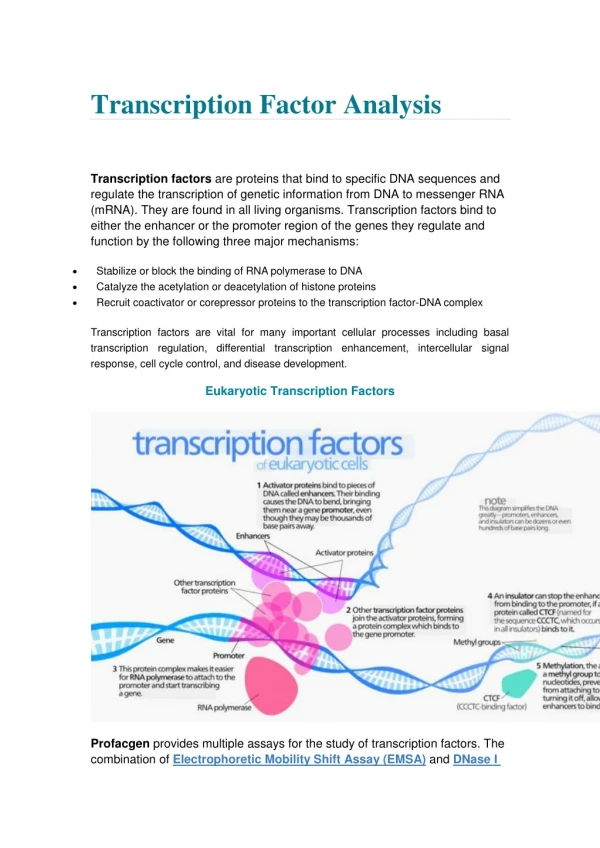
Download

1 / 29
320 likes | 465 Vues
Inferring ancestral states of the bZIP transcription factor interaction network. John Pinney. Faculty of Life Sciences University of Manchester, UK. Networks in computational biology. The genotype phenotype relationship is mediated by many inter-related biochemical networks.

E N D
Inferring ancestral states of the bZIP transcription factor interaction network John Pinney Faculty of Life Sciences University of Manchester, UK
Networks in computational biology • The genotype phenotype relationship is mediated by many inter-related biochemical networks. protein interaction gene regulation metabolismsignal transduction
Network evolution • As our knowledge of large-scale network structures improves, we can start to ask questions about the evolution of cellular systems as a whole, instead of simply looking at phylogenetic trees for individual genes. species Aspecies Bspecies Cspecies D
Network inference • We would like to be able to predict ancestral interactions based only on observations of networks from extant species. • The problem is compounded by the poor quality of high-throughput datasets (many false positives and negatives). species Aspecies Bspecies Cspecies D
Network inference by probabilistic methods • We can use a probabilistic methodology to combine multiple noisy observations of extant networks across several species. • Would like to infer probabilities for “strong” interactions between every pair of proteins in each of the least common ancestors, as well as the extant species. inferred networks observed data species Aspecies Bspecies Cspecies D
bZIP transcription factors • A useful model system for investigating methods for ancestral network inference! • Family of homo- and hetero-dimerizing proteins. • Involved in development, metabolism, circadian rhythm. • bZIP domain consists of a basic region (contacting the DNA major groove) and a leucine zipper (LZ) mediating dimerization specificity.
bZIP transcription factors • The different sub-families of bZIP proteins are known to have broadly conserved interactions with each other. GD Amoutzias et al. (2007)Mol Biol Evol24:827-835
bZIP interactions • The relative strengths of pairwise interactions between bZIP proteins have been measured experimentally for human and yeast. • In addition, the relatively simple biophysics of the coiled-coil interaction means that strong interactions can be predicted reliably from sequence data alone. JRS Newman, AE Keating (2003)Science300:2097-2101 (Darker colours show stronger interactions) JH Fong, AE Keating, M Singh (2004)Genome Biol5:R11
Danio Teleost Fugu Vertebrate Chordate Human Ciona Genomic data • Using sets of bZIP proteins from four chordate genomes, we construct a Maximum Likelihood phylogeny for the gene family with PAML. • The software by Fong et al. can be used to predict interactions between the LZ regions for the extant genomes. The scores for each pair of proteins will be our “observations” of the networks
Reconciling gene and species trees • To keep the analysis as simple as possible, we need to decide on a fixed set of proteins at each ancestral species. • This can be done by “reconciling” our gene phylogeny with the known species tree using the NOTUNG software. D Durand, BV Halldorsson, B Vernot (2006)J Comp Biol13:320-335
From gene trees to interaction trees • The model of network evolution is greatly simplified by converting to an alternative view, considering all possible interactions within a tree.
From an interaction tree to a probabilistic model • Our probabilistic graphical model of network evolution is based directly on the interaction tree. • Binary nodes represent the presence or absence of each potential interaction. • Continuous nodes are added to represent observations of interactions in extant species (our interaction scores).
Probabilistic model parameters • There are two different processes to consider in parametrising the model: • How are protein interactions re-wired as sequences evolve? • How are the observed data related to the real extant networks? false positives and negatives introduced network re-wiring species Aspecies Bspecies Cspecies D
Estimating rates of network re-wiring • It is difficult to construct a general model for gain and loss of interactions as a protein interaction network evolves. • For the bZIP network, we can estimate probabilities of gain and loss of interactions using the experimental data for human proteins. Both loss and gain of interactions are well described by logistic functions of the sum of evolutionary distances. probability P(loss of a strong interaction) loss of strong interaction d2 d1 P(gain of a strong interaction) gain of strong interaction d1 d2 d1 + d2
Adding noise to the input data • The parsimony approach might be expected to work well in cases with good quality observed data. • However, real interaction datasets are often extremely noisy. We can simulate this situation by adding Gaussian noise with different variances to the input scores. = 0 = 10 = 20 (Human input data shown)
ROC curves: Vertebrata (noise added to inputs) • As expected, the parsimony method quickly fails when the data quality falls. • The probabilistic inference method is much more robust to poor quality data, as it combines evidence across all species.
Using probabilistic inference to clean noisy interaction data • The probabilistic inference method offers a principled way to combine cross-species interaction data of various types. • This could be very useful in improving interaction predictions in extant species.
Conclusions • First successful reconstruction of ancestral interaction networks. • Parsimony method is only appropriate if input data are reliable. • Probabilistic inference works and is more robust to noisy data. • Also, probabilistic method can be used to clean up protein networks by combining cross-species data in an evolutionary context. • We hope to be able to extend this approach to model the evolution of more general classes of protein-protein interaction networks.
Acknowledgements David Robertson Magnus Rattray Grigoris Amoutzias Brian Holden Amelie Veron (Muenster) Mona Singh and Jessica Fong (Princeton)
Network inference by maximum parsimony • One straightforward method to infer ancestral networks would be to use the principle of maximum parsimony. • We calculate the minimal number of changes to the network during evolution that explain the observed data. inferred networks observed data species Aspecies Bspecies Cspecies D
Network inference using maximum parsimony • The PARS algorithm can be used to infer ancestral states of the interaction tree that are maximally parsimonious. • Interaction gains are weighted more highly than losses, as in the Bayesian approach. Interaction lost Interaction gained 1 gain, 3 losses 3 losses BG Mirkin, TI Fenner, MY Galperin, EV Koonin (2003)BMC Evol Biol3:2
Danio Teleostei Fugu Vertebrata Chordata Human Ciona Validation of inferred networks • We can also use Maximum-Likelihood methods to infer probability distributions for sequences at each of the least common ancestors. • The software by Fong et al. can then be used to predict interactions between the LZ regions for the ancestors.
Predicting interactions using sequence inference • The phylogenetic analysis software CODEML is used to infer probabilities for each amino acid at each sequence position for all nodes in the gene tree. • Sampling from these distributions allows us to predict the strength of the interaction between each pair of proteins from the same ancestral species. 90% probability of strong interaction (calibrated using human experimental data) P1 X 1000 samples P2
X Summary of methods for ancestral network inference • Gold standard: ML sequence reconstruction + sequence-based prediction • Current best method: Maximum Parsimony using PARS algorithm • New method: Inference over probabilistic model of network evolution
bZIP interactions • In addition, the relatively simple biophysics of the coiled-coil interaction means that strong interactions can be predicted reliably from sequence data alone. (70% sensitivity at 92% specificity) JH Fong, AE Keating, M Singh (2004)Genome Biol5:R11 CNC, lgMAF, smMAF families
Example: genomic data for human Darker colours show stronger predictions of interaction.
bZIP transcription factors • Gene duplication has played a major role in the evolution of the bZIP family. domain structures
Estimating error rates for predicted networks • Using the experimental human data, we can calculate the probability of a pair of proteins having a strong interaction as a function of their sequence-based interaction score.