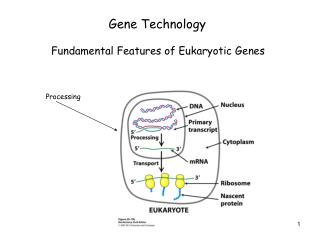
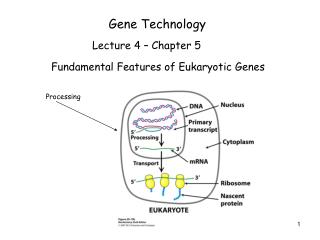
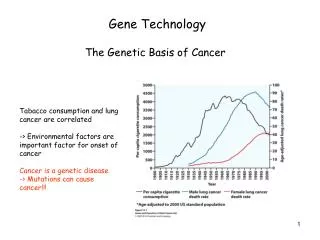
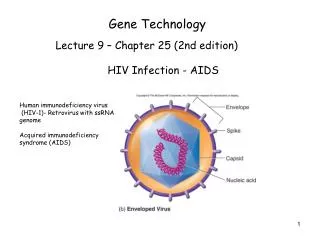
Gene Technology
Recombinant DNA Techniques – Basic tools – part 2. Screening of libraries DNA sequencing PCR. Gene Technology. A New Tool Box for Recombinant DNA – part 1. Invitro mutagenesis -> Engineering of Proteins and regulatory elements New tools for subcloning. Gene Technology.


Gene Technology
E N D
Presentation Transcript
Recombinant DNA Techniques – Basic tools – part 2 • Screening of libraries • DNA sequencing • PCR Gene Technology A New Tool Box for Recombinant DNA – part 1 • Invitro mutagenesis -> Engineering of Proteins and regulatory elements • New tools for subcloning
Gene Technology Screening of Libraries -> Identification of gene of interest • Hybridization -> screening on nucleic acid level • Requirement -> “Probe” • Methods: 1. Plaque hybridization (Phage library) • 2. Colony hybridization (bacterial library) • 2. Phenotypic -> screening on protein level • Requirements -> full length gene expressed • Methods: 1. High throughput screening (enzymatic activity screening) • 2. Antibody screening
Gene Technology Screening of Libraries -> Identification of gene of interest • Hybridization -> screening on nucleic acid level • Probe: • Small stretches of nucleic acid with a known sequence called an oligonucleotide • Single stranded • Detects specific nucleotide sequences in unknown nucleic acid samples • labeled Probe
Gene Technology Screening of Libraries -> Identification of gene of interest Blotting -> Transfer of nucleic acids to a filter (part of the hybridization procedure)
Gene Technology Screening of Libraries -> Identification of gene of interest Plaque Hybridization Colony Hybridization
Gene Technology Southern Blotting and Northern Blotting Southern Blotting: Target -> DNA Probe -> DNA -> used for detection of gene fragments Northern Blotting: Target -> RNA Probe -> DNA -> used for detection of transcription (mRNA) level replaced by Microarray and real-time PCR
Gene Technology Analysis of DNA • Electrophoresis -> fragment sizes • Hybridization and probes -> identification of fragment within a pool of DNA (library) or a larger fragment (chromosome) • Sequencing -> identification of DNA sequence of a DNA fragment • Polymerase Chain Reaction -> invitro amplification of a DNA fragment
Gene Technology DNA Sequencing according to Sanger Polymerase reaction -> with low amount of dideoxyribonucleotides (ddNTP) -> when ever ddNTP incorporated growing chain terminated
Gene Technology DNA Sequencing according to Sanger Largest fragments 3’ 5’ Smallest fragments
Gene Technology DNA Synthesis Chemical Synthesis: reaction 3’-> 5’ (no phosphate on 5’-end) -> oligonucleotides (ssDNA fragments in range between 15 – 120 bp) Used for: -> primer for sequencing or PCR (ssDNA) -> linker for cloning (dsDNA) -> assembly of whole genes (dsDNA)
Gene Technology Polymerase Chain Reaction (PCR) 1993 Kary B Mullis received the Nobel Prize in Chemistry • Specific amplification of DNA • Involves a denaturing, priming (annealing), and extension cycle • 30 cycles are sufficient for detection of DNA • Can be used to detect disease or infectious agents
Gene Technology Polymerase Chain Reaction (PCR) Annealing of primer dependent on Tm of primer -> annealing temperature 2-5 degree below Tm Extention temperature depend on polymerase -> some engineered ones are better at 68° C. In general Taq polymerase best at 72° C.
PCR Reaction mix: • Primers (15 – 30 bp) • Nucleotides (A,T, G,C) • Buffer -> Mg 2+ • Target DNA (around 10 ng) • Taq Polymerase (from Thermus aquaticus -> thermostable) Fidelity: -> rate of misincorporation -> in DNA replication : 1 in 109 nucleotides (proof reading) -> in PCR (Taq polymerase) : 1 in 2x104 nucleotides Contamination -> Target DNA normal around 10ng -> in principle PCR from one molecule possible
Gene Technology Polymerase Chain Reaction (PCR) 1st Cycle Result after 30 cycles -> just DNA between the primers amplified
Gene Technology Polymerase Chain Reaction (PCR)
Gene Technology Polymerase Chain Reaction (PCR) • Used for: • Detection of infectious diseases -> viral, bacterial infections,.. • Cloning of genes • Modifications of genes -> mutations • Screening of libraries • Detection of sex of a fetus • Forensic technology • Detection of transcription level -> real-time PCR
Gene Technology Polymerase Chain Reaction (PCR) Modification of the ends for directed cloning of PCR product:
Gene Technology Recombinant Organisms used in DNA technology • Modified bacteria and fungi • Cell lines of higher eukaryotic organisms • Transgenic plants • Transgenic animals • (Viruses)
Gene Technology Some important protein products generated by recombinant DNA technology
A New Tool Box for Recombinant DNA – part 1 Gene Technology • Invitro mutagenesis -> Engineering of Proteins and regulatory elements • New tools for subcloning
Mutagenesis Mutagenesis -> change in DNA sequence -> Point mutations or large modifications In vitro mutagenesis: -> Point mutations • Substitution: change of one nucleotide (i.e. A-> C) • Insertion: gaining one additional nucleotide • Deletion: loss of one nucleotide
Approaches for Invitro Mutagenesis -> site-directed mutagenesis -> point mutations in particular known area result -> library of wild-type and mutated DNA (site-specific) not really a library -> just 2 species -> random mutagenesis (Directed Evolution) -> point mutations in all areas within DNA of interest result -> library of wild-type and mutated DNA (random) a real library -> many variants -> screening !!! if methods efficient -> mostly mutated DNA
Gene Technology Site-directed Mutagenesis -> mainly done by PCR (with Pfu) -> only one mutation at specific location introduced -> requires knowledge of DNA sequence
Gene Technology Random Mutagenesis -> mainly done by PCR (error-prone PCR) -> Many mutations randomly incorporated -> no knowledge of DNA sequence or function required -> generate a library
Gene Technology Error-prone PCR -> PCR with low fidelity !!! Achieved by: - Increased Mg2+ concentration - Addition of Mn2+ - Not equal concentration of the four dNTPs - Use of dITP - Increasing amount of Taq polymerase (Polymerase with NO proof reading function)
Gene Technology New tools for subcloning -> Recombination Advantage: -> no restriction enzymes and no ligase are needed -> much faster 1. Cloning by use of Topoisomerase I DNA topoisomerase I -> functions as both a restriction enzyme and a ligase. Its biological role is to cleave and rejoin DNA during replication. This enables fast ligation of DNA sequences with compatible ends. After only 5 minutes at room temperature, the ligation is complete and ready for transformation into E. coli. 2.In-fusion Cloning of PCR products Create 15 bps overhangs between PCR product and vector -> allows inframe cloning with up to 90 % efficiency -> cloning of PCR product into vector done by invitro recombination -> takes only 15-30 minutes for the recombination. 3. Cloning by conjugation and recombinantion in E. coli Swap of fragment from one vector to another by the use of a lambda recombinase
Gene Technology Cloning by use of Topoisomerase I
Gene Technology In-fusion Cloning of PCR products
Gene Technology Cloning by conjugation and recombinantion in E. coli