Download
1 / 47
470 likes | 697 Vues
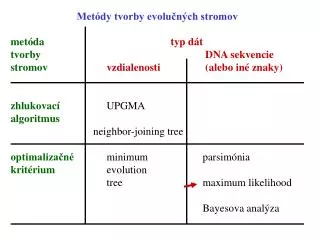
Metódy tvorby evolučných stromov. metóda t yp dát tvorby DNA sekvencie stromov vzdialenosti (alebo iné znaky) zhlukovací UPGMA algoritmus neighbor-joining tree optimalizačné minimum parsimónia kritérium evolution tree maximum likelihood

E N D
Metódy tvorby evolučných stromov metóda typ dát tvorby DNA sekvencie stromovvzdialenosti (alebo iné znaky) zhlukovací UPGMA algoritmus neighbor-joining tree optimalizačné minimum parsimónia kritérium evolution tree maximum likelihood Bayesova analýza
Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) Vierohodnosť evolučného stromu (L) je pravdepodobnosť, že sa vyvinú pozorované sekvencie DNA pri danej topológii stromu a pri danom evolučnom modeli. Cieľom metódy najväčšej vierohodnosti je nájsť strom s čo najväčšou hodnotou L. Pri tejto metóde sa zostrojí východiskový strom parsimonickou metódou alebo metódou najbližšieho suseda a potom sa prehľadávajú blízke stromy a hľadá sa pravdepodobnejší strom než ten, ktorý máme k dispozícii. Je možné aj prehľadanie všetkých možných stromov ale to, podobne ako pri parsimonických stromoch je časove náročné a realistické len pri menšom počte sekvencií. Metóda je časovo náročná. Chybný evolučný model môže viesť k chybným výsledkom.
A G T C Modely zmeny (evolúcie) sekvencií DNA Parametre, ktoré vplývajú na evolúciu sekvencií: - frekvencie báz - typy substitúcie (tranzície, tranzverzie) - heterogenita rýchlosti substitúcií puríny pyrimidíny Klasické substitučné modely: Jukes-Cantor (1969) Kimura 2 parameter (1980) Felsenstein (1981) Hasegawa, Kishino & Yano (1985) General time-reversible model (Lavane et al. 1984) Každý model vychádza z iných predpokladov
a a a a A A G G a a T T C C a A G a a a a T C a Substitučné modely JC – rovnaké rýchlosti substitúcie; rovnaké frekvencie báz ti b K2P – dve rôzne rýchlosti substitúcie; rovnaké frekvencie báz a a tv a Zvyšujúci sa počet parametrov modelu a b F81 – rovnaké rýchlosti substitúcie; nerovnaké frekvencie báz b A G a HKY – dve rôzne rýchlosti substitúcie; nerovnaké frekvencie báz a a a T C b a A G c GTR – 6 rôznych rýchlostí substitúcie; nerovnaké frekvencie báz e b d T C f
Funkcia gama distribúcie ()f(r) rýchlosti substitúcie na pozíciách sekvencie DNA • – parameter tvaru, 1 – funkcia má tvar L, 1 – funkcia má zvonovitý tvar – parameter škály proporcia pozícií f(r) Yang, Z., 1996, Trends Ecol. Evol. 11: 367-372. rýchlosť substitúcií (r)
W W W X Y Z Y X X Z Z Y strom 1 strom 2 strom 3 Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) Niektoré z možných stromov Sekvencia W:A C G C GT TG G GSekvencia X:A C G C GT TG G GSekvencia Y:A C G C A ATGA ASekvencia Z:A C A C A G G GA A
T T A G T A T G T G T A strom 1 strom 2 strom 3 Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) Niektoré z možných stromov Sekvencia W:A C G C GT TG G GSekvencia X:A C G C GT TG G GSekvencia Y:A C G C A ATGA ASekvencia Z:A C A C A G G GA A
T T T T T T T T A A A A G G G G T T A G T T A T A G A C G T G A Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) Niektoré z možných evolučných ciest k jednému zo stromov (rôzne topológie)
T T A G ATGC ATGC ATGC Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) Možné evolučné cesty k jednému zo stromov počet evol. ciest k jednému stromu / 1 pozícia: (počet stavov)(počet uzlov) = (počet stavov)(počet taxónov -1) = 43 = 64
T T A G T G G Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) Vierohodnosť (likelihood) jednej evolučnej cesty L = L(koreň/root) x P L(konáre/branches) závisí od konkrétneho modelu
T T A G Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) Vierohodnosť (likelihood) jedného stromu z jednej pozície DNA sekvencie L(strom z jednej pozície) = S L(všetky evolučné cesty k stromu z jednej pozície) = L(strom 1) + L(strom 2) + L(strom 3) + … + L(strom 64) - stromy rovnakej topológie čo do taxónov na koncoch vetiev ale s inými nukleotidmi v uzloch
W X Y Z Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) Vierohodnosť (likelihood) jedného stromu z celej DNA sekvencie L(strom za celú sekvenciu) = P L(strom za určitú pozíciu)
Metóda najväčšej vierohodnosti (maximum likelihood (ML)method) ML analýza začína stromom vytvoreným napr. NJ a potom sa prehľadávajú blízke stromy a hľadajú sa tie, ktoré majú vyššiu vierohodnosť. Pre väčšie dátové súbory heuristická analýza ML je počítačovo veľmi náročná, navyše podpora jednotlivých clades by mala byť overená metódou bootstrapu, čo násobí potrebný čas na analýzu. Pokiaľ ML analýza nájde lokálne optimum, ešte to neznamená, že ide zároveň aj o globálne optimum v stromovom priestore.
Metóda najväčšej vierohodnosti (maximum likelihood, ML) • ML analýza zahŕňa: • testovanie evolučných modelov pre daný dátový súbor, výpočet skóre (log likelihood scores) vzhľadom na model a dáta - PAUP-výber vhodného modelu na základe log likelihood scores podľa určitých testovacích kritérií - MODELTEST- výpočet (hľadanie) najpravdepodobnejšieho stromu podľa vybraného modelu – PAUP • podrobné a jednoduché inštrukcie pre výpočet: http://www.rhizobia.co.nz/phylogenetics/modeltest.html MODELTEST (http://darwin.uvigo.es/software/modeltest.html) • program na porovnanielog likelihood scores testovaných modelov, výber vhodného evolučného modelu pre daný dátový súbor- príručka: modeltest3.6.pdf • Posada D and Crandall KA 1998. Modeltest: testing the model of DNA substitution. Bioinformatics 14 (9): 817-818.Posada D and Buckley TR. 2004. Model selection and model averaging in phylogenetics: advantages of the AIC and Bayesian approaches over likelihood ratio tests. Systematic Biology 53: 793-808
modely sú vyjadrené parametrami: - frekvencia báz, - typy substitúcie a ich rýchlosti, - homogenita/heterogenita mutačných rýchlostí na rôznych pozíciách, - podiel invariabilných pozícií rýchlosti mutácií a(A-C), b(A-G), c(A-T), d(C-G), e(C-T), f(G-T)
1. nexus súbor s pridaným textovým blokom (príkazmi) na hodnotenie 56 evolučných modelova výpočet skóre (výpočet v PAUP-e)
1. nexus súbor s pridaným textovým blokom (príkazmi) na hodnotenie 56 evolučných modelova výpočet skóre (výpočet v PAUP-e) výpočet počiatočného stromu (NJ)
1. nexus súbor s pridaným textovým blokom (príkazmi) na hodnotenie 56 evolučných modelova výpočet skóre (výpočet v PAUP-e) výpočet log likelihood scores pre NJ strom vzhľadom na dáta a model
Lscores 1... výpočet log likelihood score pre NJ strom pre daný model, vyjadrený: nst...number of substitution types (1-6)base...frekvencia bázrates...heterogenita mutačnej rýchlosti na rôznych pozíciáchshape...alfa parameter funkcie gamma distribúciepinv...proporcia invariabilných pozícií
2. načítanie nexus súboru v programe PAUP a výpočetskóre (log likelihood scores) pre jednotlivé modely
2. výpočetlikelihoodskóre (log likelihood scores) pre jednotlivé modely -> model.scores
2. výpočetlikelihoodskóre (log likelihood scores) pre jednotlivé modely -> model.scores
3. načítanie súboru model.scores v programe MODELTESTvzájomné testovanie modelov na základe log likelihood scores, výber vhodného modelu podľa dvoch testovacích kritérií
3. načítanie súboru model.scores v programe MODELTEST - definovanie cesty a adresára, kde je uložený spúšťací súbor Modeltest3.6.exe(cd – change directory) - príkaz na spustenie programu MODELTEST a výpočet -> výstup v súbore *.outfile
4. súbor *.outfileobsahuje vzájomné testovanie modelov na základe log likelihood scores, výber vhodného modelu podľa dvoch testovacích kritérií – AIC (Akaike information criterion), hLRT (hierarchical likelihood ratio test) hLRT – vzájomné porovnávanie dvoch modelovAIC – simultánne porovnanievšetkých modelov nie model s najvyšším skóre, ale s najoptimálnejším (t.j. ak pridanie parametrov, zvýšenie komplexity len nepatrne zvýši skóre, vyberá sa menej komplexný model) niekedy navrhnú odlišné modely, je na užívateľovi, ktorý si vyberie, preferované AIC
4. súbor *.outfileobsahuje vzájomné testovanie modelov na základe log likelihood scores, výber vhodného modelu podľa dvoch testovacích kritérií – AIC, hLRT
4. súbor *.outfile špecifikované parametre vybraného modelu pre ML výpočet lset...špecifikácia vybraného modelu Base...frekvencia báz A, C, G (T)Nst... počet typov substitúciíRmat... rýchlosti mutácií a(A-C), b(A-G), c(A-T), d(C-G), e(C-T), f(G-T)Rates...funkcia gama distribúcie (rýchlosť mutácií na nukleotidových pozíciách)Shape...parameter tvaru fumkciePinvar...proporcia invariabilných pozícií
5. nexus súbor s pridanou špecifikáciou vybraného modelu a príkazmi na ML výpočet
MrMTgui http://www.genedrift.org/mtgui.php
http://www.bio.utexas.edu/faculty/antisense/garli/Garli.html http://www.nescent.org/informatics/download.php?software_id=4
program TCS http://darwin.uvigo.es/software/tcs.html Clement M, Posada D and Crandall K. 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9(10): 1657-1660 tvorba siete haplotypov (cpDNA dáta) - štatistická parsimónia Templeton, A.R., Crandall, K.A., Sing, C.F., 1992. A cladistic analysis of the phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132, 619–633Templeton, A.R., 1998. Nested clade analyses of phylogeographic data: testing hypotheses about gene flow and population history. Mol. Ecol. 7, 381–397.
TCS Connection limit: percentuálne 90-95% počet mutačných krokov Gaps = missing Gaps = 5th state
TCS uloženie siete haplotypov ako - *.gml súbor ako - *.ai súbor
TCS log súbor: