Fibrous Proteins
Fibrous Proteins. UNIT I: Protein Structure and Function. Overview. Collagen & elastin are examples of common, well-characterized fibrous proteins that serve structural functions in the body


Fibrous Proteins
E N D
Presentation Transcript
Fibrous Proteins UNIT I: Protein Structure and Function
Overview • Collagen & elastin are examples of common, well-characterized fibrous proteins that serve structural functions in the body • E.g., collagen & elastin found as components in skin, connective tissue, blood vessel walls, & sclera and cornea of eye. • Each fibrous protein exhibits special mechanical properties, resulting from unique structure, obtained by specific aa’s combined into regular 2º structural elements. • In contrast to globular proteins, whose shapes result from complex interactions b/w 2º, 3º, and sometimes 4º elements.
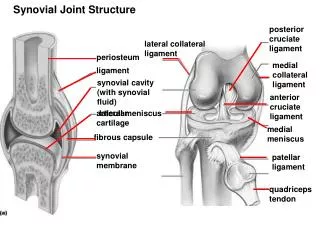
II. Collagen • Is most abundant protein in human body • A typical molecule is long, rigid, in which 3 polyps (α-chains) wound around one another in a rope-like triple-helix. • Although found throughout the body, their types and organization dictated by structural role collagen plays in a particular organ. • In some tissues, collagen may be dispersed as a gel that give support to structure, as in extracellular matrix or the vitreous humor of eye. • In other tissues, collagen may be bundled in tight, parallel fibers that provide great strength, as in tendons. • In cornea, collagen is stacked so as to transmit light with a minimum of scattering. • Collagen of bone occurs as fibers arranged at an angle to each other to resist mechanical shear from any direction
The cornea is made up of many layers of collagen arranged in a very regular pattern. These layers of collagen are called the stromal lamellae
The molecular arrangement of collagen and hydroxyapatite crystals in compact bone. (a) Collagen fibers overlap adjacent fibers as they repeat every 680 Å. Hole zones are areas of low density bone collagen and overlap zones are areas of very high density bone collagen. (b) Hydroxyapatite crystalsare arranged in layers within each fiber, resembling overlapping bricks
Figure 4.1 Triple-stranded helix of collagen.
A. Types of collagen • Collagen superfamily of proteins include > 20 collagen types, as well as additional proteins that have collagen-like domains. • The 3 polyp. α-chains are held together by H-bonds b/w the chains. • Variations in aa sequence of α-chains result in structural components that are ~ the same size (~ 1000 aa’s long), but slightly different properties. • These chains are combined to form various types of collagen found in tissues. E.g., most common collagen, type I, contains 2 chains α1 and 1 chain α2 (α12α2), whereas type II collagen contains 3 α1 chains (α13). • The collagens can be organized into 3 groups, based on location and function in the body.
Figure 4.2 The most abundant types of collagen.
Figure 4.3 Collagen fibrils at right have a characteristic banding pattern, reflecting the regularly staggered packing of the individual collagen molecules in the fibril.
1. Fibril-forming collagens: • Types I, II, and III are fibrillar collagens, & have rope–like structure • In EM, these linear polymers of fibrils have characteristic banding patterns, reflecting regular staggered packing of individual collagen molecules in fibril. • Type I collagen fibers are found in supporting elements of high tensile strength (e.g., tendon & cornea) • Fibers formed from type II are restricted to cartilaginous structures. • Fibrils derived from type III are prevalent in more distensible tissues, e.g., blood vessels
2. Network-forming collagens • Types IV and VII form 3 D mesh, rather than fibrils. E.g., type IV molecules assemble into a sheet or meshwork that constitutes a major part of basement membranes • Basement membranes are thin, sheet like structures that provide mechanical support for adjacent cells, & function as a semipermeable filtration barrier for macromolecules in organs such as kidney & lung
Figure 4.4. Electron micrograph of a polygonal network formed by association of collagen type IV monomers.
3. Fibril-associated collagens • Types IX and XII bind to surface of collagen fibrils, linking them to one another and to other components in extracellular matrix
B. Structure of collagen. • Amino acid sequence: collagen is rich in Pro & Gly, both important in formation of triple-stranded helix. • Pro facilitates formation of helical conformation of each α-chain because its ring causes “kinks” in peptide chain. • Gly, smallest aa, is found in every 3rd position of the polyp chain. It fits into restricted spaces where the 3 chains of the helix come together • Gly residues are part of a repeating sequence, -Gly-X-Y-, X: frequently Pro, Y: often hydroxyproline or hydroxylysine • Thus, most α-chain can be regarded as a polyp whose seq can be represented as (-Gly-X-Y-)333.
Figure 4.5 Amino acid sequence of a portion of the a1-chain of collagen. [Note: Hyp is hydroxyproline and Hyl is hydroxylysine.]
2. Triple helical structure: unlike most globular proteins that are folded into compact structures, collagen, a fibrous protein, has an elongated, triple helical structure that places many of its aa’s side chains on surface of the triple helical molecule - This allows bond formation b/w exposed R-groups of neighboring collagen monomers aggregation into fibers
3. Hydroxyproline & hydroxylysine: • Collagen contains hyp & hyl, not present in most other proteins • These residues result from hydroxylation of some Pro & Lys residues after incorporation into polyp chains • Hydroxylation is an e.g. of post-translational modification • Hydroxylation of Pro is important in stabilizing triple-helical structure of collagen, it maximizes interchain H-bond formation
Figure 4.6 Hydroxylation of prolyl residues of pro-α-chains of collagen by prolyl hydroxylase.
3. Glycosylation: • Hydroxyl group of hyl residues of collagen may be enzymatically glycosylated • Most commonly, glucose & galactose are sequentially attached to polyp chain prior to triple-helix formation
Figure 4.7 Formation of a collagen fibril.
Figure 4.7 Formation of a collagen fibril. (Continued from the previous page)
C. Biosynthesis of collagen • Polyp precursors of collagen molecule are formed in fibroblasts (or in related osteoblasts of bone and chondroblasts of cartilage) and secreted into extracellular matrix. • After enzymatic modification, mature collagen monomers aggregate & become cross-linked to form collagen fibrils
Formation of pro-α-chains: • Collagen is one of many proteins that normally function outside of cells • Like most proteins produced for export, newly synthesized polyp precursors of α-chains contain a special aa sequence at their N-terminus, acts as a signal that polyp being synthesized is destined to leave cell • Signal sequence facilitates binding of ribosomes to rER, & directs passage of polyp chain into cisternae of rER • Signal sequence is rapidly cleaved in ER to yield a precursor of collage = pro- α-chain
2. Hydroxylation • Pro- α-chains processed by a number of enzymatic steps within lumen of rER while polyp is being synthesized • Pro & Lys residues found in Y-position of –Gly-X-Y- sequence can be hydroxylated to form hyp and hyl residues • Hydroxylation reactions require molecular oxygen and the reducing agent vitamin C (ascorbic acid), without which hydroxylating enzymes, prolyl hydroxylase & lysyl hydroxylase, are unable to function.
In case of ascorbic acid deficiency (so, lack of prolyl and lysyl hydroxylation), collagen fibers cannot be crosslinked greatly decreasing tensile strength of assembled fiber • One resulting deficiency disease is scurvy. • Patients with ascorbic acid deficiency also often show bruises on limbs as a result of subcutaneous extravasation of blood (capillary fragility) Figure 4.8. The legs of a 46-year-old man with scurvy.
3. Glycosylation: some hydroxylysine residues are modified by glycosylation with glucose or glucosyl-galactose 4. Assembly and secretion: • After hydroxylation & glycosylation, pro-α-chains form pro-collagen, a precursor of collagen that has a central region of triple helix flanked by non-helical amino- and carboxyl-terminal extensions called propeptides
Formation of procollagen begins with formation of interchain disulfide bonds b/w C-terminal extensions of pro-α-chains, this brings three α-chains into an alignment favorable for helix formation • Procollagen molecules translocated to Golgi, packaged in secretory vesicles vesicles fuse with CM release of procollagen molecules into extracellular space
5. Extracellular cleavage of procollagen molecules: • After release, pro-collagen molecules are cleaved by N- and C-procollagen peptidases, which remove terminal propeptides, releasing triple-helical collagen molecules 6. Formation of collagen fibrils: • Individual collagen molecules spontaneously associate to form fibrils. • They form ordered, overlapping, parallel array, with adjacent collagen molecules arranged in a staggered pattern, each overlapping its neighbor by a length ~ three-quarters of a molecule.
7. Cross-link formation: • Fibrillar array of collagen molecules serves as a substrate for lysyl oxidase. This extracellular enz oxidatively deaminates some lysyl and hyl residues in collagen. Reactive aldehydes that result (allysine and hydroxyallysine) can condense with lysyl or hyl residues in neighboring collagen molecules to form covalent cross-links • This cross-linking is essential for achieving tensile strength necessary for proper functioning of connective tissue. Therefore, any mutation that interferes with ability of collagen to form cross-linked fibrils almost certainly affects stability of collagen
Figure 4.9 Formation of cross-links in collagen.
D. Degradation of collagen • Normal collagens highly stable molecules, having half-lives as long as several months. However, connective tissue is dynamic & is constantly being remodeled, often in response to growth or injury • Breakdown of collagen fibrils is dependent on proteolytic action of collagenases, which are part of a large family of matrix metalloproteinases. • For type-I collagen, cleavage site is specific, generating 3-quarter and 1-quarter length fragments. These fragments are further degraded by other matrix proteinases to their constituent aa’s
E. Collagen diseases • Defects in any one of the many steps in collagen fiber synthesis can result in a genetic disease involving an inability of collagen to form fibers properly, provide tissues with needed tensile strength normally provided by collagen • > 1000 mutations have been identified in 22 genes coding for 12 of the collagen types
1. Ehlers-Danlos syndrome (EDS) • Is a heterogeneous group of generalized connective tissue disorders result from inheritable defect in metabolism of fibrillar collagen molecule • EDS can result from deficiency of collagen processing enzymes (e.g., lysyl-hydroxylase deficiency or pro-collagen peptidase deficiency), or from mutations in aa sequences of collagen types I,III, or V. • Most clinically important mutations found in the gene for type III collagen
Collagen containing mutant chains is not secreted, and is either degraded or accumulated to high levels in intracellular compartments • As collagen type III is important component of arteries, potentially lethal vascular problems occur • Although collagen III is only a minor component of collagen fibrils in skin, for unknown reasons, EDS patients also show defects in collagen type I fibrils stretchy skin & loose joints
2. Osteogenesis imperfecta (OI): • A.k.a brittle bone syndrome, also a heterogeneous group of inherited disorders distinguished by bones that easily bend and fracture • Retarded wound healing & a rotated and twisted spine leading to a “humped-back” appearance are common features • Type I OI is called osteogenesis imperfecta tarda. Presents in early infancy with fractures secondary to minor trauma, and may be suspected if prenatal ultrasound detects bowing or fractures of long bones
Figure 4.11. Lethal form of osteogenesis imperfecta in which the fractures appear in utero, as revealed by this radiograph of a stillborn fetus.
Type II OI, osteogenesis imperfecta congenita, is more severe, and patients die in utero or in the neonatal period of pulmonary hypoplasia • Most patients with severe OI have mutations in the gene for either proα-1- or pro-α2 chains of type I collagen. • Most common mutations cause substitution of single aa’s with bulky side chains for Gly residues that appear as every 3rd aa in the triple helix • Structurally abnormal pro-α-chains can prevent folding of protein into a triple-helical conformation
III. Elastin • Elastin is a connective tissue protein with rubber-like properties. • Elastic fibers composed of elastin & glycoprotein microfibrils are found in lungs, walls of large arteries, & elastic ligaments. They can be stretched to several times their normal length, but recoil to their shape when stretching force is relaxed
A. Structure of elastin • Insoluble protein polymer synthesized from a precursor, tropoelastin: a linear polymer ~ 700 aa’s that are primarily small & non-polar (e.g., Gly, Ala, Val) • Elastin is also rich in Pro & Lys, but contains only little hyp & no hyl • Tropoelastin is secreted by the cell into extracellular space. There it interacts with specific glycoprotein microfibrils, e.g., fibrillin, which function as scaffold onto which tropoelastin deposited • Mutations in fibrillin gene Marfan’s syndrome
Some lysyl side chains of tropoelastin polyps oxidatively deaminated by lysyl oxidase allysine residues • 3 allysyl side chains + 1 unaltered lysyl side chain from same or neighboring polyps form a desmosine cross-link • This produces elastin- an extensively interconnected, rubbery network that can stretch and bend in any direction when stressed connective tissue elasticity
Figure 4.13. Elastin fibers in relaxed and stretched conformations.
Role of α1-antitrypsin in elastin degradation • α1-antitrypsin : blood & other body fluids contain a protein, α1-AT (a.k.a α1-antiproteinase), inhibits a number of proteolytic enz’s (a.k.a proteases or proteinases) that hydrolyze & destroy proteins. • Originally named α1-antitrypsin as it inhibits activity of trypsin (a protease synthesized as trypsinogen by pancreas) • α1-AT comprises > 90% of α1-globulin fraction of normal plasma.
α1-AT has important physiologic role of inhibiting neutrophil elastase: a powerful protease released into extracellular space & degrades elastin of alveolar walls, as well as other structural proteins in a variety of tissues • Most α1-AT found in plasma is synthesized by several tissues, including monocytes & alveolar macrophages, which may be important in prevention of local tissue injury by elastase
2. Role of α1-AT in lungs • In normal lung, alveoli are chronically exposed to low levels of neutrophil elastase released from activated and degenerated neutrophils • This proteolytic activity can destroy elastin in alveolar walls if unprotected by inhibitory action of α1-AT, the most important inhibitor of neutrophil elastase • As lung tissue cannot regenerate, emphysema results from destruction of connective tissue of alveolar walls
Figure 4.14 Destruction of alveolar tissue by elastase released from neutrophils.
3. Emphysema resulting from α1-AT deficiency • In US ~ 2-5% of patients with emphysema are predisposed to disease by inherited defects in α1-AT. • A number of different mutations in α1-AT gene are known to cause a deficiency of this protein, but one single purine base mutation (GAG AAG: lys for glu at position 342) is clinically the most widespread • An individual must inherit 2 abnormal α1-AT alleles to be at risk for development of emphysema • In a heteozygote, levels of α1-AT are sufficient to protect alveoli from damage
A specific α1-AT Met is required for binding of inhibitor to its target proteases. Smoking causes oxidation & subsequent inactivation of that Met residue, thereby rendering the inhibitor powerless to neutralize elastase. • Smokers with α1-AT deficiency have a considerably elevated rate lung destruction & poorer survival rate than non-smokers with the deficiency • Deficiency of elastase inhibitor can be reversed by weekly administration of α1-AT. The α1-AT diffuses from blood into lung, where it reaches therapeutic levels in the fluid surrounding the lung epithelial cells