Download

1 / 20
210 likes | 384 Vues
Imaging of Biological Molecules in Solution by Small Angle X-ray Scattering.

E N D
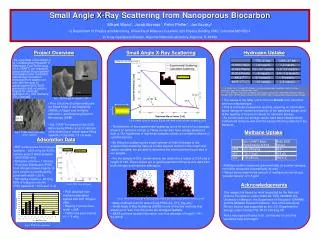
Imaging of Biological Molecules in Solution by Small Angle X-ray Scattering. Mark J. van der Woerd1, Donald Estep2, Simon Tavener2, F. Jay Breidt3, Stefan Sillau3, James Bieman4, Michelle Strout4, Christopher Wilcox4 Sanjay Rajopadhye4 and Karolin Luger1. 1Department of Biochemistry & Molecular Biology, 2Department of Mathematics, 3Department of Statistics, 4Department of Computer Science, Colorado State University, Fort Collins, CO 80523.
The size of the problem Approx 240 x 200 x 60 m 7 x 2.5 x 2.5 mm 10 x 10 x 3.5 nm Factor 5 * 1013 Factor 1017 We are visualizing objects on the order of 10-9 m, guiding wavelength and technique
What motivates us? • We are interested in understanding the function of the machinery that enables life • Function is closely linked to structure • The machinery consists of DNA and proteins, among other things • We need to know the structure of individual biological molecules (protein, DNA, RNA) alone, and of their complexes in order to begin to understand function
How do we determine structure? • Traditionally there are three methods: • Protein Crystallography • Nuclear Magnetic Resonance (NMR) Spectroscopy • Modeling • We are now pursuing Small Angle X-ray Scattering on biological systems in solution (SAXS).
Log (I) S (function of q) How does SAXS work? Radially symmetric scattering pattern Incident X-rays q Sample in solution, typically 10 ml, inside a quartz container. All particles in solution will scatter X-rays, since electrons (present in all atoms) are interacting with the X-rays. How? Structural information
Advantages of the technique: • Don’t need to make (protein) crystals – this is a very time consuming and complicated process; this is a solution method • Experimentally very simple, experiments can be done in a few minutes • Can determine the global shape of large complexes of molecules, which is difficult to do with other techniques suitable for structure determination, while it is biologically very interesting.
Data processing consists of: • Signal correction (subtracting background, correcting for incident beam, time- and concentration-dependent corrections) • Either ab initio model building from raw data • Or using existing models as ‘puzzle pieces’ • Or a combination of these methods Generally build a model by some method, generate a ‘calculated’ scattering pattern from the model and compare with experimental outcome. Iterate to minimize the differences.
Example: Nucleosome Assembly Protein - 1 • It helps in compacting DNA into nucleosomes, the first step in the process of folding DNA into chromosomes • It helps DNA to ‘slide’ so the ‘correct piece’ can be exposed and used at the right time • How does it work? • It interacts with… what?
Log (I) S (function of q) Test the method with NAP-1 alone Spheres approach Build a model of spheres that has the appropriate size and shape so that the predicted solution scattering pattern closely matches the experimental data. This is an ab initio approach because there is no prior information put into generating the model.
Log (I) S (function of q) Test the method with NAP-1 alone Puzzle piece approach Acquire a model from another investigation and use it as a ‘puzzle piece’, try to see if one or more pieces combined can explain the experimental data. This method is particularly useful when we study large complexes of known structures. In this case the model was obtained by means of X-ray crystallography (spheres are representations of atoms).
New Method Development • We need reliable, scientifically transparent methods to interpret scattering data. • New method development involves Biochemistry, Physics, Mathematics, Statistics, Computer Science. • Next couple of slides are an outline of plans for ongoing and future research
Biophysics • Is it possible to develop a method which can be used to include or exclude models that are deemed good or bad? • Example: protein molecules must be internally sound, they do not contain ‘voids’. This is similar to asking ‘how does a protein fold’?
Mathematics • It is implicitly assumed in our model that all molecules tumble rapidly and all molecular orientations are equally likely in our sample. • This may not be correct and we would like to test systems that do not incorporate completely randomly oriented molecules. • How does this affect the scattering pattern (if at all)? Need alternative description?
Statistics • Suppose you had two models that both seem reasonable, could you assign a quality descriptor to the models and tell which is ‘best’, i.e. which fits the experimental data the best? • Possible approach: use of maximum likelihood methods.
Computer Science • The generation of possible models that fit the experimental data is very time consuming: what are efficient methods to speed up the programs? • The proposed process of image reconstruction from scattering data is complex: what is a good way to write a program suite that works well and can be easily maintained?
Application • We have our preliminary results: can we extend into the unknown? • How can we best assure that the results are scientifically sound? • To which parts of the nucleosome does NAP-1 bind and how does this affect the formation of new or change of existing nucleosomes?
Funding: HHMI NIH Center for Interdisciplinary Mathematic & Statistics – by extension Offices of the Dean and Vice President for Research Lot of help and patience: Drs. Michal Hammel and Greg Hura (LBNL) ALS (LBNL) for beam time Acknowledgments