Download

1 / 21
380 likes | 790 Vues
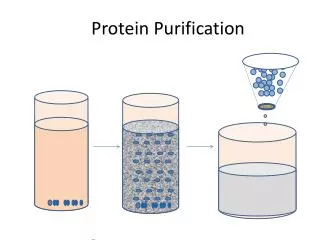
Protein purification. Chroma tography – Mikhail Tsvett (1901) pioneered the technique while attempting to separate yellow and red pigments from green leaves.

E N D
Protein purification Chromatography – Mikhail Tsvett (1901) pioneered the technique while attempting to separate yellow and red pigments from green leaves
If you don’t have the gene that encodes the protein but you have a source, you may want to purify the protein for any of the following reasons: • The purified protein can be used to determine the amino acid sequence. • The purified protein can be used to make antibodies. • The purified protein can be analyzed and identified by mass spectroscopy. • If you have the gene that encodes the protein, you may want to purify the protein for other reasons: • Pure proteins are required for structural analysis.(x-ray crystallography and NMR spectroscopy). • Pure proteins are required to study enzyme function. • Pure proteins can be used to determine what other proteins or molecules • they might interact with. • Pure proteins are needed for studies of protein function (e.g. Are there regulatory subunits? Is it phosphorylated? Is the protein regulated by its interactions with other proteins? Etc.)
So you want to make a pure protein? • An entire protein ? • b) A domain from a mosaic protein ? If yes, don’t need to worry about limits * Need to worry about limits
Learn all you can before beginning MSA can often give you ideas for deciding on construct limits Even better if there’s some structural information!
If multiple sequence alignments do not help and there isn’t any structural info, try secondary structure prediction ..but try several starts and stops (primers are cheap!) http://www.igb.uci.edu/tools/scratch/
Lots of output! Near bottom, look for “mRNA Sequence” – That’s the accession # you want (RefSeq record)
Also American Type Culture Collection and Research Genetics/Invitrogen Integrated Molecular Analysis of Genomes and their Expression
Expression and purification of YFP Bacterial expression system Advantages – Easy, great over-expression, low protease activity, no post- translational modifications Disadvantages - Protein solubility, lack of post- translational modifications Eukaryotic expression system Advantages - Protein solubility, post-translational modifications Disadvantages - Expense, low yield, proteases Isolate protein from native source Advantages – Protein solubility, authenticity Disadvantages - Expense/effort, yield, slaughter-houses Waring blenders, Gross!(Gt) Hierarchy – Bacteria, Yeast, SF9, Hela, native tissue
Before starting, confirm that you can make a significant quantity of soluble protein. Small scale solubility experiments are very important and typically will involve varying inducer concentration, expression temperature, expression construct, etc. Each protein is unique – must exploit differences Particular affinities GST, 6xHis, antibodies Solubility (NH4)2SO4, PEG precip. Charge ion exchange Hydrophobicity hydrophobic chromatography Size gel exclusion Iso-electric point iso-electric focusing Thermal stability alter temp.
Standard methods Express protein in frame with an affinity tag – often tag is removable with a protease. Common tags: 6xHis, GST, CaM, MBP. Use affinity chromatography for first step! electron coordination bonds Imidazole Nitrilotriacetic acid pH 7.4 Kirkegaard & Perry Laboratories, Inc
If the affinity tag is removable, go back over column and collect flow-through (or digest on the column). Ion exchange chromatography (what is the theoretical pI of your protein?) DiEthylAminoEthane (DEAE), CarboxyMethyl (CM), Quaternary amine, Sulfonic acid. http://www.proteinchemist.com/tutorial/iec.html These functional groups are charged over a broad pH range. Why would that be desirable?
Anion exchange (example ion exchange) Anion #2 ( Cl- ) Anion #1 ( protein ) Na+ + + - - pH=6 Na+ Cl- + + - - + Na+ + Cl- + + YFP YFP - - + Na+ + + Cl- + - - + + Na+ Cl- + + Bind (Low salt) Elute (High salt)
Run a 20 x (column volume) linear gradient and collect fractions 500mM NaCl Linear gradient (also step) Trp, Tyr, Phe, disulfides 50mM NaCl example chromatogram Run SDS-PAGE of fractions to decide which to pool (sacrifice yield for purity?)
Stronger and higher resolution ion exchange media (Q, SP) may be employed to separate proteins that were not baseline separated with weak ion exchange step. Some proteins, usually larger proteins, can bind to both anion and cation exchange matrices – change pH to enhance interaction. Electrostatic potential mapped onto a molecular surface Q column SP column
Gel exclusion chromatography – Separates proteins by size. Your protein should elute at the proper volume for its expected Mr. Want a nice, symmetric peak in the chromatogram. Small proteins “see” a bigger volume than do large proteins
Some other chromatographic techniques Salting out – Proteins precipitate differentially in the presence of (NH4)2SO4 or polyethylene glycol - It’s probably worth trying Hydrophobic – Load proteins onto phenyl sepharose in presence of ~1.5M (NH4)2SO4 and run decreasing [(NH4)2SO4] gradient. More hydrophobic elutes later. Isoelectric focusing – Electrophorese protein in matrix containing pH gradient. When the protein reaches that pH where it has no net charge it ceases to migrate. Retrieve protein from matrix.
What one does in practice is discover a purification protocol. It depends on the level of purity required, but no matter what the vendors say, there is no one step purification to homogeneity (in my experience!)