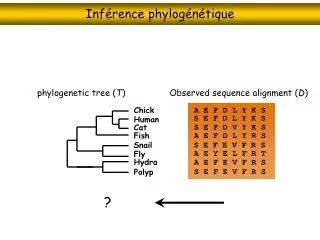
Inférence phylogénétique
570 likes | 669 Vues
Inférence phylogénétique. phylogenetic tree ( T ). Observed sequence alignment ( D ). Chick. A E F D L Y K S. S E F D L Y K S. Human. S E F D V Y K S. Cat. Fish. A E F D L Y R S. Snail. S E F E V F R S. A E Y E L F R T. Fly. Hydra. A E F E V F R S. S E F E V F R S. Polyp. ?.

Inférence phylogénétique
E N D
Presentation Transcript
Inférence phylogénétique phylogenetic tree (T) Observed sequence alignment (D) Chick A E F D L Y K S S E F D L Y K S Human S E F D V Y K S Cat Fish A E F D L Y R S Snail S E F E V F R S A E Y E L F R T Fly Hydra A E F E V F R S S E F E V F R S Polyp ?
Principes généraux d'une méthode d'inférence phylogénétique Deux aspects indépendants à considérer : • Le critère: Se définir un score S(T,D) permettant de mesurer le 'fit' d'un arbre T, sachant les données D (séquences alignées) • La méthode de recherche: Choisir parmi tous les arbres possibles de celui ayant le meilleur score
Critère 1 : Maximum de parcimonie Pour un arbre donné, le score est le nombre minimal de changements nécessaires pour expliquer l’alignement phylogenetic tree (T) Observed sequence alignment (D) Chick A E F D L Y K S S E F D L Y K S Human S E F D V Y K S Cat Fish A E F D L Y R S Snail S E F E V F R S A E Y E L F R T Fly Hydra A E F E V F R S S E F E V F R S Polyp
Critère 1 : Maximum de parcimonie Pour un arbre donné, le score est le nombre minimal de changements nécessaires pour expliquer l’alignement phylogenetic tree (T) Observed sequence alignment (D) Chick A E F D L Y K S S E F D L Y K S Human D E S E F D V Y K S Cat Fish A E F D L Y R S Snail S E F E V F R S E A E Y E L F R T Fly Hydra A E F E V F R S S E F E V F R S Polyp
Critère 1 : Maximum de parcimonie Pour un arbre donné, le score est le nombre minimal de changements nécessaires pour expliquer l’alignement phylogenetic tree (T) Observed sequence alignment (D) Chick A E F D L Y K S S E F D L Y K S Human S E F D V Y K S Cat Fish A E F D L Y R S Snail S E F E V F R S A E Y E L F R T Fly Hydra A E F E V F R S S E F E V F R S Polyp
Critère 1 : Maximum de parcimonie Pour un arbre donné, le score est le nombre minimal de changements nécessaires pour expliquer l’alignement phylogenetic tree (T) Observed sequence alignment (D) Chick A E F D L Y K S S E F D L Y K S L V Human V L S E F D V Y K S Cat Fish A E F D L Y R S Snail S E F E V F R S L V V A E Y E L F R T Fly Hydra A E F E V F R S S E F E V F R S Polyp Une convergence : deux évolutions indépendantes vers L Une réversion : chez le chat, un retour à l'état ancestral (V) Convergences et réversions : homoplasies
Critère 1 : Maximum de parcimonie Pour un arbre donné, le score est le nombre minimal de changements nécessaires pour expliquer l’alignement phylogenetic tree (T) Observed sequence alignment (D) Chick A E F D L Y K S S E F D L Y K S Human V L S E F D V Y K S Cat Fish A E F D L Y R S V L Snail S E F E V F R S L A E Y E L F R T Fly V L Hydra A E F E V F R S S E F E V F R S Polyp
Maximum de parcimonie Deux étapes de minimisation : • Pour un arbre donné, minimisation du nombre de changements nécessaires pour expliquer l’alignement • Choix parmi tous les arbres possibles de celui ayant le plus petit nombre de changements
Trouver l'arbre de Maximum de parcimonie méthode naïve • prendre chaque arbre l'un après l'autre • pour chaque arbre, calculer le score de parcimonie • conserver le (ou les) meilleurs arbres
Trouver l'arbre de Maximum de parcimonie méthode naïve • prendre chaque arbre l'un après l'autre • pour chaque arbre, calculer le score de parcimonie • conserver le (ou les) meilleurs arbres • problème: combinatoire trop élevée
Heuristiques de recherche de topologies Agglomération progressive des espèces • Insertion sur toutes les branches • Calcul du nombre de changements • Choix de l’arbre le plus parcimonieux Nombre d’opérations : 3+5+7=15 (pour 105 arbres possibles) Pour 10 espèces : 63 versus 2 millions
D A E C F B C D D A F B E C C D D A F B E C B E C D D A F B E C A F B E Ordre d’agglomération des espèces • Ajouter les espèces de manière aléatoire • Répéter l’opération un grand nombre de fois • Choisir l’arbre le plus parcimonieux
X X X Z Z Y W W Y Z W Y 2(n-3) possibilités Subtree Pruning and Regrafting (SPR) 3 4 3 4 1 5 1 5 6 2 6 2 3 4 3 4 1 5 1 5 6 2 6 2 4(n-3)(n-2) possibilités Algorithme de réarrangements Réarrangementlocal ou Nearest Neighbor Interchange (NNI)
Minimums locaux : les ilôts d’arbres optimaux RÉPLICATS D’ADDITION ALÉATOIRE DE SEQUENCES ÉCHEC SUCCÈS ÉCHEC Branch Swapping Branch Swapping Branch Swapping Longueur De L’arbre Minimum Local Minimums Locaux MINIMUM GLOBAL http://bioinf.ncl.ac.uk/molsys/data/characters.ppt
The branch and bound algorithm Hendy et Penny (1982) Mathematical Biosciences, 60:133-142, 1982
Utilisation de contraintes a priori G2 G4 G3 G1 G5 G11 G9 G10 G8 G6 11 espèces : 24 106 45 espèces : 5 1064 G7
Méthode de distances • Pour chaque paire d’espèces, calculer la distance évolutive (c’est-à-dire le nombre de différences) • Choix parmi tous les arbres possibles de celui correspondant à la matrice de distances • Le critère ici est l'adéquation entre l'arbre et la matrice de distance
Cas idéal Espèce 1 CUAAAAAAAAA Espèce 2 -AGG------- Espèce 3 AA------UGC Espèce 4 AA--GGCU--C E1 E2 E3 E4 Espèce 1 0 Espèce 2 3 0 Espèce 3 5 6 0 Espèce 4 7 8 6 0
Cas idéal AAAAAAAAAAA 1 1 AAAAAAAAAAC CAAAAAAAAAA 1 2 2 4 CUAAAAAAAAA CAGGAAAAAAA AAAAAAAAUGC AAAAGGCUAAC Espèce 4 Espèce 3 Espèce 2 Espèce 1 Espèce 1 CUAAAAAAAAA Espèce 2 -AGG------- Espèce 3 AA------UGC Espèce 4 AA--GGCU--C E1 E2 E3 E4 Espèce 1 0 Espèce 2 3 0 Espèce 3 5 6 0 Espèce 4 7 8 6 0
1 1 3 3 1 a 2 c e 2 b 2 d 4 4 4 2 2 Principe des méthodes de distance E1 E2 E3 E4 Espèce 1 0 Espèce 2 3 0 Espèce 3 5 6 0 Espèce 4 7 8 6 0 Arbre #1 D(E1,E2) = a+b=3 (1) D(E1,E3) = a+e+c=5 (2) D(E1,E4) = a+e+d=7 (3) D(E2,E3) = b+e+c=6 (4) D(E2,E4) = b+e+d=8 (5) D(E3,E4) = c+d=6 (6) Plus d’équations que de variables : en général, pas de solutions (2)+(4)-(1) 2e+2c=5+6-3 e+c=4 (7) (2)–(7) a=5-4=1 (1) b=3-1=2 (4)+(5)–(6) 2b+2e=6+8-6 b+e=4 e=4-2=2 (4) c=6-2-2=2 (5) d=8-2-2=4
1 1 2 2 1 a b 2 e 0 4 c d 6 4 4 3 3 Principe des méthodes de distance E1 E2 E3 E4 Espèce 1 0 Espèce 2 3 0 Espèce 3 5 6 0 Espèce 4 7 8 6 0 Arbre #2 D(E1,E2) = a+e+b=3 (1) D(E1,E3) = a+c=5 (2) D(E1,E4) = a+e+d=7 (3) D(E2,E3) = b+e+c=6 (4) D(E2,E4) = b+d=8 (5) D(E3,E4) = c+e+d=6 (6) Plus d’équations que de variables : en général, pas de solutions (2)+(4)-(1) 2c=5+6-3 c=4 (2) a=5-4=1 a=1 (3)+(4)-(5) a+c+2e=7+6-8=5 e=0 (1) b=3-1=2 b=2 (5) d=8-2=6 d=6 (6) 4+0+6=6 10=6
d(i,k) = d(j,k) d(i,j) = d(i,k) d(j,i) = d(j,k) d(j,k) d(i,k) d(i,j) i j k j i k i j k Une matrice de distances est dite ultramétrique si et seulement si, pour toutes feuilles i,j et k, max{d(i,j), d(i,k), d(j,k)} n’est pas unique Horloge moléculaire Distances additives et ultramétriques Une matrice de distances est dite additive (d’arbre) si et seulement il existe un arbre ayant des longueurs de branches parfaitement compatibles avec la matrice Conditions des 4 points : Pour tout ensemble de 4 espèces i,j,k et l, deux des valeurs D(i,j)+D(k,l), D(i,k)+D(j,l) et D(i,l)+D(j,k) sont égales et supérieures à la troisième
AAAAAAAAAAA 1 1 AAAAAAAAAAC CAAAAAAAAAA 1 2 2 4 CUAAAAAAAAA CAGGAAAAAAA AAAAAAAAAGU AAAAGGCUAAC Espèce 4 Espèce 3 Espèce 2 Espèce 1 Espèce 1 CUAAAAAAAAA Espèce 2 -AGG------- Espèce 3 AA-------GU Espèce 4 AA--GGCU--C Une substitution multiple Substitutions multiples entraînent une sous-estimation des distances E1 E2 E3 E4 Espèce 1 0 Espèce 2 3 0 Espèce 3 4 (5) 5 (6) 0 Espèce 4 7 8 6 0 Distances non additives
Moindres carrés • Appelons P une distance correspondant à un arbre phylogénétique T • les espèces i et j, P(i,j) = Sarètes sur le chemin connectant i et j • Soit F la fonction qui mesure la qualité de l’ajustement (fit) de P à D • ou • On calcule les arètes de longueur inconnue (a, b, c, d et e, dans le cas précédent) en minimisant F par une méthode de moindre carré (least square method) • On choisit parmi tous les arbres possibles celui qui minimise F • Comme en parcimonie, deux étapes d’ajustement/minimisation
Calculer Chercher i et j pour lesquels « Dij - ui - uj» est minimal i j i vi Calculer les voisinages : vi = (Dij + ui - uj) / 2 vj = (Dij + uj - ui) / 2 j vj Calculer les distances du nouveau noeud (ij) à k : D(ij),k = (Dik + Djk - Dij) / 2 (ij) Répéter jusqu’à ce qu’il ne reste que 3 espèces Algorithme du Neighbor Joining (Saitou&Nei, 1987)
AAAAAAAAAAA 1 1 1 1.5 E1 E3 1.5 2 AAAAAAAAAAC 4.5 CAAAAAAAAAA E2 E4 1 2 2 4 CUAAAAAAAAA CAGGAAAAAAA AAAAAAAAAGU AAAAGGCUAAC Espèce 4 Espèce 3 Espèce 2 Espèce 1 Une substitution multiple E1 E2 E3 E4 Espèce 1 0 Espèce 2 3 0 Espèce 3 4 (5) 5 (6) 0 Espèce 4 7 8 6 0
AAAAAAAAAAA 1 1 AAAAAAAAAAC CAAAAAAAAAA 1 2 2 4 CUAAAAAAAAA GAAGAAAAAAA AAAGAAAAAAA AAAAGGCUAAC Espèce 4 Espèce 3 Espèce 2 Espèce 1 Trois substitutions multiples E1 E2 E3 E4 Espèce 1 0 Espèce 2 3 0 Espèce 3 3 (5) 1 (6) 0 Espèce 4 7 7 (8) 6 0 1.8 0.6 E1 E2 0.7 E4 E3 5.2 0.4 Nécessité de corriger les distances avant d’inférer l’arbre
Calcul de la distance évolutive Mutation : altération d’une molécule d’ADN (mutation ponctuelle : si un seul nucléotide est modifié) Substitution : modification de la séquence d’ADN dans l’ensemble de la population Distance évolutive = nombre de substitutions ayant eu lieu entre deux espèces (ou organismes) PRINCIPE : estimer à partir des différences observées entre deux séquences le nombre de substitutions qui se sont produites Nécessité d’un modèle d’évolution des séquences
A simple model: Jukes and Cantor Evolution operates independently and identically at each position …A A C A G T A C… Markov process - no memory - time-homogeneous - of rate G C …A A C A C T A C… C A For each substitution, draw final state with equal probability (1/3) A G …A G C A A T A C… time
t 0 Séquence S0 Séquence St + l/3*(1-qt) sites qui étaient différents Modèle de Jukes et Cantor (1969) • Toutes les substitutions sont équiprobables (par exemple si A est modifié, il y a une chance sur trois que ce soit en C, 1/3 en G et 1/3 en T) • Tous les sites ont la même probabilité de subir une substitution Soit l la fréquence de substitutions par million d'années Soit qt la fréquence des nucléotides identiques entre la séquence à l’instant t et la séquence à l’instant 0 On cherche à calculer qt+1 la fréquence des nucléotides identiques entre la séquence à l’instant t + 1 et la séquence à l’instant 0 qt+1 = (1-l)*qt sites qui restent constants
0 Séquence S1 : S0 Séquence S2 : S2t 2t Modèle de Jukes et Cantor (1969) • Toutes les substitutions sont équiprobables (par exemple si A est modifié, il y a une chance sur trois que ce soit en C, 1/3 en G et 1/3 en T) • Tous les sites ont la même probabilité de subir une substitution D : distance évolutive (nombre de substitutions) Q : p-distance (nombre de différences observées)
Modèle de Jukes et Cantor (1969) 1 0.9 0.75 0.8 0.7 0.6 0.5 p-distance (différences observées) 0.4 0.3 0.2 0.1 0 0 1 2 3 4 5 6 DJC (substitutions estimées)
Modèle de Kimura (1980) • Transition : remplacement d’une purine (A et G) par une autre ou d’une pyrimidine (C et T) par une autre. • Transversion : remplacement d’une purine (A et G) par une pyrimidine (C et T).
P : fréquence de transitions observées entre 2 séquences Q : fréquence de transversions observées entre 2 séquences Modèle de Kimura (1980) • Transition : remplacement d’une purine (A et G) par une autre ou d’une pyrimidine (C et T) par une autre. • Transversion : remplacement d’une purine (A et G) par une pyrimidine (C et T). • Les substitutions sont séparées en deux catégories : transitions et transversions. A l’intérieur de ces deux catégories, les changements sont équiprobables (même probabilité de passer de A vers C que de A vers T) • Tous les sites ont la même probabilité de subir une substitution
Modèle de Jukes et Cantor (1969) 1 0.9 0.75 0.8 0.7 (2 nucléotides) 0.50 0.6 0.5 p-distance (différences observées) 0.4 0.3 0.2 0.1 0 0 1 2 3 4 5 6 DJC (substitutions estimées)
A simple model Felsenstein 1981 Evolution operates independently and identically at each position …A A C A G T A C… Markov process - no memory - time-homogeneous - of rate G C …A A C A C T A C… C A For each substitution, draw final state from frequency vector: A G …A G C A A T A C… time A C G T F G H I K L M N P Q R S T V W Y
P : fréquence de différences observées entre 2 séquences Modèle de Felsenstein (1981) • Toutes les substitutions sont équiprobables (par exemple si A est modifié, il y a une chance sur trois que ce soit en C, 1/3 en G et 1/3 en T) • Tous les sites ont la même probabilité de subir une substitution • Les fréquences des quatre nucléotides ne sont pas obligatoirement 0.25, 0.25, 0.25 et 0.25
General Time Reversible (GTR) model • Chaque type de substitutions nucléotidiques a sa propre probabilité • Tous les sites ont la même probabilité de subir une substitution • Les fréquences des quatre nucléotides ne sont pas obligatoirement 0.25, 0.25, 0.25 et 0.25
300 250 200 150 100 50 0 Variabilité du taux de substitutions entre positions Nombre de substitutions par position Alignement de cytochrome b 2065 séquences de cytochrome b de Vertebrés
300 250 160 200 140 120 150 100 100 80 60 50 40 20 0 0 Observé Ajusté (a=0.54) Variabilité du taux de substitutions entre positions Nombre de substitutions par position Alignement de cytochrome b
Modélisation par une distribution G a : paramètre de forme
Variabilité du taux de substitutions entre positions Nei et Gojobori (1986) Jin et Nei (1990) Tamura et Nei (1993) a, paramètre de la loi G p, P, P1, P2 et Q fréquences des différences, transitions,transitions A/G, transitions CXT et transversions, respectivement f, fréquence des nucléotides (R=purines, Y=pyrimidines)
Robustesse des phylogénies : l’indice de Bremer Indice de Bremer : nombre de pas à ajouter à la longueur de l’arbre le plus parcimonieux pour casser la monophylie du clade considéré Indice de Bremer (“Bremer Decay Index”) est égal à : Le nombre de pas de l’arbre le plus parcimonieux dans lequel le clade considéré n’est pas monophylétique Moins Le nombre de pas de l’arbre le plus parcimonieux dans lequel le clade considéré est monophylétique
Robustesse des phylogénies : le test du bootstrap • Tirage avec remise de n positions parmi n positions • Construire l’arbre phylogénétique • Répéter 1) et 2) un grand nombre de fois (1000) • Analyser tous les arbres obtenus (en particulier via un arbre consensus)
C D D B D C A A A E E E F F F B C B C D A E 93% 52% 100% F B L’arbre de consensus majoritaire Etc. • On calcule la fréquence d’apparition des différents groupements d’espèces • E et F : 100% • D, E et F : 93% • A et B : 52% • A et C : 48% • C, E et F : 7% • Construire l’arbre consensus
Robustesse des phylogénies : le test du jackknife Jackknife : tirage sans remise de x% des positions originales 1 1 0 1 1 1 1 0 1 1 1 0 1 0 1 0 1 1 0 1 1 1 1 0 Le but du jackknife est de savoir si une petite fraction de l’alignement a un impact sur l’inférence phylogénétique
Robustesse des phylogénies : le test du bootstrap Le tirage avec remise de positions, en respectant l’effectif original, revient à conférer un poids aléatoire aux positions Par exemple : 2 1 0 3 0 2 1 1 1 3 0 4 0 0 1 2 2 1 1 3 0 1 0 1 3 2 0 0 (à chaque tirage, un tiers des positions ne sont pas considérés) Le but du bootstrap vise à mesurer si le signal phylogénétique en faveur d'un groupe donné est présent dans beaucoup de colonnes de l'alignement • Problèmes • Très couteux en temps calcul • Seuil de significativité (70%, 95%)
Le principe de parcimonie • Interpretation "Philosophique" • rasoir d'Occam • l'économie d'hypothèses est à la base de la science • Interprétation probabiliste • chaque fois que deux espèces ont un caractère en commun, • deux interprétations sont possibles: • 1- caractère hérité de l'ancêtre commun • 2- convergence (acquisition indépendante) • Maximum de Parcimonie <=> 1 est plus probable que 2
Le principe de parcimonie mis en défaut D'après l'interprétation probabiliste, toutes les fois où les convergences sont plus probables que les caractères dérivés partagés, le principe du Maximum de Parcimonie est inconsistant.