Download

1 / 48
480 likes | 607 Vues
Explore the timeline, properties, and implications of the human genome project, including gene recognition difficulties and protein complexities. Discover insights from the sequencing and analysis of the human genome data.

E N D
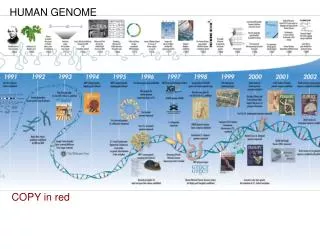
The Human Genome The International Human Genome Consortium Initial sequencing and analysis of the human genome Nature, 409, February 15, 860-921 (2001) Venter et al. (Celera) The Sequence of the Human Genome Science, 291, February 16, 1304-1351 (2001) HC LEE January 8, 2002 Computational Biology Lab National Central University
1984 to 1986 – first proposed at US DOE meetings • 1988 – endorsed by US National Research Council • creation of genetic, physical and sequence maps of • the human genome • parallel efforts in key model organisms: bacteria, yeast, • worms, flies and mice; • develop of supporting technology • ethical, legal and social issues (ELSI) • 1990 – Human Genome Project (NHGRI) • Later – UK, France, Japan, Germany, China
Completed sequences 1995 – First complete bacterial genomes 2002 – About 35 bacterial genomes; 0.5-5 Mb; hundreds to 2000 genes 1996 April – Yeast (Saccharomyces cerevisiae) 12 Mb, 5,500 genes 1998 Dec. -Worm (Caenorhabditis elegans) 97 Mb, 19,000 genes 2000 March - Fly (Drosophila melanogaster) 137 Mb, 13,500 genes 2000 Dec. - Mustard (Arabidopsis thaliana) 125 Mb, 25,498 genes 2000 June – Human (Homo sapiens) 1st rough draft 2001 Feb 15/16 – Human, “working draft” 3000 Mb, 35,000~40,000 genes
IHGCS paper Nature, 409, February 15, 860-921 (2001)
Celera paper Science, 291, February 16, 1304-1351 (2001)
Sequencing BAC: Bacterial Artificial Chromosome clone Contig: joined overlapping collection of sequences or clones.
C-value paradox • C-value paradox: Genome size does • not correlate well with organismal • complexity. • Human Homo sapiens 3000 Mb • Yeast S. cerevisiae 12 Mb • Amoeba dubia 600,000 Mb • Genomes can contain a large quantity • of repetitive sequence, far in excess of • that devoted to protein-coding genes
Global properties • Pericentromeric and subtelomeric regions of chromosomes filled with large recent transposable elements • Marked decline in the overall activity of transposable elements or transposons • Male mutation rate about twice female • most mutation occurs in males • Recombination rates much higher in distal regions of chromosomes and on shorter chromosome arms • > one crossover per chromosome arm in each meiosis
Important features of Human proteome • 30,000–40,000 protein-coding genes • Proteome (full set of proteins) more complex than those of invertebrates. • pre-existing components arranged into a richer architectures. • Hundreds of genes seem to come from horizontal transfer from bacteria • Dozens of genes seem to come from transposable elements.
Human proteome is complex • Gene codes proteins (also RNAs) • Number of genes does not reflect complexity of organism Org’nism no. genes no. proteins Worm 20,000 ~20,000 Fly 13,500 >>20,000 Human ~40,000 >>100,000
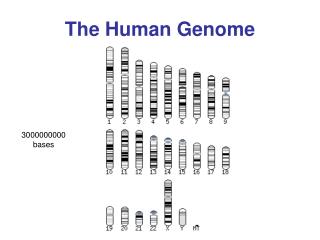
Human genome content • The Human Genome • Total length 3000 Mb • ~ 40,000 genes (coding seq) • Gene sequences < 5% • Exons ~ 1.5% (coding) • Introns ~ 3.5% (noncoding) • Intergenic regions (junk) > 95% • Repeats > 50%
Gene codes proteins (also RNAs) (transcription & translation) Procaryotes (single cell): one gene, one protein Eucaryote (multicell): gene = intron + exon; one gene, many proteins
Fig 35a Size distributions of exons in Human, Worm and Fly. Human have shorter exons.
Fig 35c Size distributions of intons in Human, Worm and Fly. Human have longer introns.
Gene recognition • Coding region and non-coding region have different sequence profiles • coding region is “protected” from mutation and is less random • Gene recognition by sequence alignment • Gene prediction by Hidden Markov Model trained by set of known genes • Many genes are homologs – similar in vastly different organisms
Gene recog’n difficult for Human • Easy for procaryotes (single cell) – one gene, one protein • More difficult for eukaryotes (multicell) – one gene, many proteins • Very difficult for Human – short exons separated by non-coding long introns
Genes predicted in Human Genome Int’l Consortium Celera known genes 14,882 17,764 novel genes 16,896 21,350 Total 31,778 39,114
Two predictions disagree “…predicted transcripts collectively contain partial matches to nearly all known genes, but the novel genes predicted by both groups are largely non-overlapping.” John B. Hogenesch, et al Cell, Vol. 106, 413–415 August 24, 2001
Global properties with evolutionary implications • Long-range variation in GC content not random • CpG islands protected by genes • Genetic and physical distance non-linear • > 50% genome composed of repeats
Standard deviation 15 times wider than random distrib’n GC-rich and GC-poor regions have different biological properties, such as gene density, composition of repeat sequences, correspondence with cytogenetic bands and recombination rate.
GC content in introns (exons) vs introns (exons) length.
Fig 14 CpG islands CpG islands and genes are correlated. CpG dinucleotides are methylated; methyl-CpG steadily mutate to TpG. Hence CpG is greatly under-represented in human DNA. Except in CpG islands near genes.
Fig 15 recomb rate (distal) Recombination rate vs Physical position from centromere of genes. Rate higher in distal regions.
Fig 16 recomb rate (short arm) Recombination rate higher on shorter chromosome arms
The genome mutates and copies itself • 50%, probably much more, of genome composed of repeats • Many traces of repeats obliterated by mutation • Lower organisms may have longer genomes • Five types of repeats • transposable elements; processed pseudogenes; simple k-mer repeats; segmental duplications (10-300 kb); (large) blocks of tandemly repeated sequences
Fig 17 transposables Interspersed repeats: fixed transposable elements copied to non-homologous regions. Total 45% Classes of transposable elements. LINE, long interspersed element. SINE short interspersed element.
Fig 21 Genes are sometimes protected from repeats Two regions of about 1 Mb on chromosomes 2 and 22. Red bars, interspersed repeats; blue bars, exons of known genes. Note the deficit of repeats in the HoxD cluster, which contains a collection of genes with complex, interrelated regulation.
Tab 14 SSR content Simple sequence (k-mers) repeats: SSR
Fig 32b Mosaic patterns of duplications. For each region top horizon line: segment of sequence (100–500 kb) with interchromosomal (red) and intrachromosomal (blue) duplications displayed. Lower lines with a distinct colours: separate sequence duplication. y axis: per cent nucleotide identity. b. An ancestral region from Xq28 that has contributed various 'genic' segments to pericentromeric regions.
Fig 32a An active pericentromeric region on chromosome 21.
Fig 32c c. A pericentromeric region from chromosome 11.
Fig 32d d. A subtelomeric region from chromosome 7p.
Fig 33 Finished HG has 1.5% interchromosomal 2% intrachromosomal segmental duplications. The duplications are 10–50 kb long and highly homologous. Structure in similarity may indicate that interchromosomal duplications occurred in a punctuated manner.
Human Proteome • Number of human genes (~40,000) only twice that of worm or fly • Many more transcripts (combination of exons in one gene) • Many more proteins, perhaps >> 100,000 • Most proteins are still homologs of non-human proteins • Homologs (from a common ancestor gene) • orthologs – derived through speciation • paralogs: derived through duplication
Completed eukaryotic proteomes Human Fly Worm Yeast Mustard weed Identified genes 32,000 13,338 18,266 6,144 25,706 Annotated domain families 1,262 1,035 1,014 861 1,010 Distinct domain architectures 1,695 1,036 1,018 310 -
Fig 38 distribution of homologs Distribution of homologues of predicted human proteins
Simplified cladogram (relationship tree) of the 'many-to-many' relationships of classical nuclear receptors. Triangles indicate expansion within one lineage; bars represent single members. Numbers in parentheses indicate the number of paralogues in each group.
Fig 42 domain accretion Domain accretion in chromatin proteins in various lineages before the animal divergence, in the apparent coelomate lineage and the vertebrate lineage are shown using schematic representations of domain architec- tures (not to scale). Asterisks, mobile domains that have participated in theaccretion. Species in which a domain architecture has been identified are indicated (Y, yeast; W, worm; F, fly; V, vertebrate).
Fig 45 domain expansion Lineage-specific expansions of domains and architectures of transcription factors
Conserved segments in human and mouse genome Colour code: Mouse genome
Applications to medicine and biology • Disease genes • human genomic sequence in public databases allows rapid identification of disease genes in silico • Drug targets • pharmaceutical industry has depended upon a limited set of drug targets to develop new therapies • now can find new target in silico • Basic biology • basic physiology, cell biology…
The next steps • Finishing the human sequence • Developing the IGI (integrated gene index) and IPI (protein) • Large-scale identification of regulatory regions • Sequencing of additional large genomes • mouse, super-rice, pig, fish… • Completing the catalogue of human variation • Single nucleotide polymorphism • nasal and throat cancer… • From sequence to function