Download

1 / 43
610 likes | 1.21k Vues
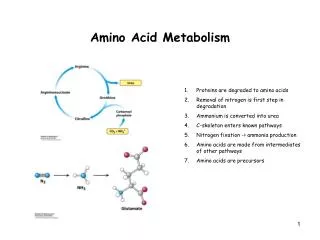
AMINO ACID METABOLISM. Jana Novotná Department of the Medical Chemistry and Biochemistry The 2nd Faculty of Medicine, Charles Univ. Amino acid structure. The 20 common amino acids of proteins. BODY PROTEINS. 250 – 300 g/day. Degradation. Proteosynthesis. NONPROTEIN DERIVATIVES

E N D
AMINO ACID METABOLISM Jana Novotná Department of the Medical Chemistry and Biochemistry The 2nd Faculty of Medicine, Charles Univ. Downloaded from www.pharmacy123.blogfa.com
Amino acid structure Downloaded from www.pharmacy123.blogfa.com
The 20 common amino acids of proteins Downloaded from www.pharmacy123.blogfa.com
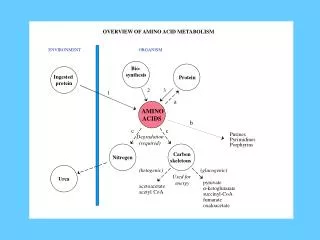
BODY PROTEINS 250 – 300 g/day Degradation Proteosynthesis NONPROTEIN DERIVATIVES Porphyrins Purines Pyrimidines Neurotransmitters Hormones Komplex lipids Aminosugars Digestion AMINO ACIDS DIETARY PROTEINS GLYCOLYSIS KREBS CYCLE Transamination Conversion (Carbon skeleton) UREA NH3 GLUCOSE ACETYL CoA CO2 KETONBODIES Metabolic relationship of amino acids Downloaded from www.pharmacy123.blogfa.com
Enzymes cleaving the peptide bond Endopeptidases – hydrolyse the peptide bond inside a chain: pepsin, trypsin, chymotrypsin Exopeptidases – split the peptide bond at the end of a protein molecule: aminopeptidase, carboxypeptidases Dipeptidases pepsin (pH 1.5 – 2.5) – peptide bond derived from Tyr, Phe, bonds between Leu and Glu trypsin (pH 7.5 – 8.5) – bonds between Lys a Arg chymotrypsin (pH 7.5 – 8.5) – bonds between Phe a Tyr Downloaded from www.pharmacy123.blogfa.com
Essential amino acids in humans Lysine Methionine Threonine Phenylalanine Tryptophan Arginine* Histidine* Isoleucine Leucine Valine Downloaded from www.pharmacy123.blogfa.com *Required to some degree in young growing period and/or sometimes during illness.
Non-essential and nonessential amino acids in humans Can be formed from a-keto acids by transamination and subsequent reactions. Alanine Asparagine Aspartate Glutamate Glutamine Glycine Proline Serine Cysteine (from Met*) Tyrosine (from Phe*) * Essential amino acids Downloaded from www.pharmacy123.blogfa.com
+ NH4+ O O C C COO- COO- R R deamination transamination NH2 NH2 CH CH COO- COO- R R NH3+ oxidative decarboxylation CH2 R + CO2 General reactions of amino acid catabolism Downloaded from www.pharmacy123.blogfa.com
The fate of the amino group during amino acid catabolism Downloaded from www.pharmacy123.blogfa.com
Transamination reaction The first step in the catabolism of most amino acids is removal of a-amino groups by enzymes transaminases or aminotransferases All aminotransferases have the same prostethic group and the same reaction mechanism. The prostethic group is pyridoxal phosphate (PPL), the coenzyme form of pyridoxine (vitamin B6) Downloaded from www.pharmacy123.blogfa.com
Biosynthesis of amino acid: transamination reactions amino acid1+a-keto acid2 amino acid2 +a-keto acid1 Keto-acid + Glutamate Pyridoxal phosphate (PLP)- dependent aminotransferase Amino acid + a-Ketoglutarate Downloaded from www.pharmacy123.blogfa.com
Active metabolic form of vitamin B6 Downloaded from www.pharmacy123.blogfa.com
Mechanism of transamination reaction: PPL complex with enzyme accept an amino group to form pyridoxamine phosphate, which can donate its amio group to an a-keto acid. Downloaded from www.pharmacy123.blogfa.com
Allamino acids except threonine, lysine, and proline can be transaminated Transaminases are differ in their specificity for L-amino acids. The enzymes are named for the amino group donor. Downloaded from www.pharmacy123.blogfa.com
Clinicaly important transaminases Alanine-a-ketoglutarate transferase ALT (also called glutamate-pyruvate transaminase – GPT) Aspartate-a-ketoglutarate transferase AST (also called glutamate-oxalacetate transferase – GOT) Important in the diagnosis of heart and liver damage caused by heart attack, drug toxicity, or infection. ALT Downloaded from www.pharmacy123.blogfa.com
Glucose-alanine cycle Alanine plays a special role in transporting aminogroups to liver. Ala is the carrier of ammonia and of the carbon skeleton of pyruvate from muscle to liver. The ammonia is excreted and the pyruvate is used to produce glucose, which is returned to the muscle. Downloaded from www.pharmacy123.blogfa.com According to D. L. Nelson, M. M. Cox :LEHNINGER. PRINCIPLES OF BIOCHEMISTRY Fifth edition
Glutamate releases its amino group as ammonia in the liver The amino groups from many of the a-amino acids are collected in the liver in the form of the amino group of L-glutamate molecules. • Glutamate undergoes oxidative deamination catalyzed by L-glutamate dehydrogenase. • Enzyme is present in mitochondrial matrix. • It is the only enzyme that can use either NAD+ or NADP+ as the acceptor of reducing equivalents. • Combine action of an aminotransferase and glutamate dehydrogenase referred to as transdeamination. Downloaded from www.pharmacy123.blogfa.com
Ammonia transport in the form of glutamine Excess ammonia is added to glutamate to form glutamine. Glutamine synthetase Glutamine enters the liver and NH4+ is liberated in mitochondria by the enzyme glutaminase. Ammonia is remove by urea synthesis. Downloaded from www.pharmacy123.blogfa.com
Relationship between glutamate, glutamine and a-ketoglutarate NH3 NH3 E E a-ketoglutarate glutamate glutamine NH3 NH3 E A. Glutamate dehydrogenase E + + + + H2O glutamate a-ketoglutarate NH3 NADH NAD+ To urea cycle From transamination reactions B. Glutamine synthetase (liver) ADP ATP E + glutamine glutamate NH3 C. Glutaminase (kidney) E + + H2O Downloaded from www.pharmacy123.blogfa.com glutamate NH3 glutamine
A. Oxidative deamination Amino acids + FMN + H2O E L-amino acid oxidase + a-keto acids FMNH2 + NH3 O2 catalse E H2O + O2 FMN H2O2 B. Nonoxidative deamination serine threonine E Serin-threonin dehydratase E pyruvate + NH3 a-ketoglutate + NH3 Oxidative deamination • L-amino acid oxidase produces ammonia and a-keto acid directly, using FMN as cofactor. • The reduced form of flavin must be regeneratedby O2 molecule. • This reaction produces H2O2 molecule which is decompensated by catalase. Is possible only for hydroxy amino acids Downloaded from www.pharmacy123.blogfa.com
Amino acid metabolism and central metabolic pathways • 20 amino acids are converted to 7 products: • pyruvate • acetyl-CoA • acetoacetate • a-ketoglutarate • succynyl-CoA • oxalacetate • fumarate Downloaded from www.pharmacy123.blogfa.com
Glucogenic Amino Acids formed:a-ketoglutarate, pyruvate,oxaloacetate, fumarate, or succinyl-CoA Alanine Serine Cysteine Glycine Threonine Tryptophan Methionine Valine Glutamine Glutamate Proline Histidine Aspartate Asparagine Arginine Phenylalanine Tyrosine Isoleucine Downloaded from www.pharmacy123.blogfa.com
Ketogenic Amino Acids formed acetyl CoA or acetoacetate Lysine Leucine Downloaded from www.pharmacy123.blogfa.com
Both glucogenic and ketogenic amino acids formed:a-ketoglutarate, pyruvate,oxaloacetate, fumarate, or succinyl-CoA in addition to acetyl CoA or acetoacetate Isoleucine Threonine Tryptophan Phenylalanine Tyrosine Downloaded from www.pharmacy123.blogfa.com
Alanine Serine Cysteine Threonine The C3 family: alanine, serine, cysteine and threonine are converted to pyruvate Pyruvate Downloaded from www.pharmacy123.blogfa.com
Asparagine Aspartic acid Oxalacetate The C4 family: aspartate and asparagine are converted into oxalacetate Downloaded from www.pharmacy123.blogfa.com
The C5 family: several amino acids are converted into a-ketoglutarate through glutamate Glutamine a-ketoglutarate Proline Arginine Histidine Downloaded from www.pharmacy123.blogfa.com
Interconversion of amino acids and intermediates of carbohydrate metabolism and Krebs cycle Downloaded from www.pharmacy123.blogfa.com
Metabolism of some selected amino acids Downloaded from www.pharmacy123.blogfa.com
Serine biosynthesis from glycolytic intermediate 3-phosphoglycerate Downloaded from www.pharmacy123.blogfa.com Copy from:http://themedicalbiochemistrypage.org/amino-acid-metabolism.html
Glycine biosynthesis from serine Reaction involves the transfer of the hydroxymethyl group from serineto the cofactor tetrahydrofolate (THF), producing glycine and N5,N10-methylene-THF. Downloaded from www.pharmacy123.blogfa.com Copy from:http://themedicalbiochemistrypage.org/amino-acid-metabolism.html
Glycine oxidation to CO2 Glycine produced from serine or from the diet can also be oxidized by glycine decarboxylase (also referred to as the glycine cleavage complex, GCC) to yield a second equivalent of N5,N10-methylene-tetrahydrofolate as well as ammonia and CO2. Downloaded from www.pharmacy123.blogfa.com Copy from:http://themedicalbiochemistrypage.org/amino-acid-metabolism.html
Cysteine and methionine are metabolically related The sulfur for cysteine synthesis comes from the essential amino acid methionine. SAM Condensation of ATP and methionine yield S-adenosylmethionine (SAM) SAM serves as a precurosor for numerous methyl transfer reactions (e.g. the conversion of norepinephrine to epinenephrine). Downloaded from www.pharmacy123.blogfa.com
Cysteine synthesis * Conversion of homocysteine back to Met. N5-methyl-THF is donor of methyl group. *folate + vit B12 • Conversion of SAM to homocysteine. • Condensation of homocysteine with serine to cystathione. • Cystathione is cleavaged to cysteine. Downloaded from www.pharmacy123.blogfa.com Copy from:http://themedicalbiochemistrypage.org/amino-acid-metabolism.html
Homocystinuria Genetic defects for both the synthase and the lyase. Missing or impaired cystathionine synthase leads to homocystinuria. High concentration of homocysteine and methionine in the urine. Homocysteine is highly reactive molecule. Disease is often associated with mental retardation, multisystemic disorder of connective tissue, muscle, CNS, and cardiovascular system. Downloaded from www.pharmacy123.blogfa.com
Biosynthesis of Tyrosine from Phenylalanine Phenylalanine hydroxylase is a mixed-function oxygenase: one atom of oxygen is incorporated into water and the other into the hydroxyl of tyrosine. The reductant is the tetrahydrofolate-related cofactor tetrahydrobiopterin, which is maintained in the reduced state by the NADH-dependent enzyme dihydropteridine reductase Downloaded from www.pharmacy123.blogfa.com
Phenylketonuria Hyperphenylalaninemia- complete deficiency of phenylalanine hydroxylase (plasma level of Phe raises from normal 0.5 to 2 mg/dL to more than 20 mg/dL). The mental retardation is caused by the accumulation of phenylalanine, which becomes a major donor of amino groups in aminotransferase activity and depletes neural tissue of α-ketoglutarate. Absence of α-ketoglutarate in the brain shuts down the TCA cycle and the associated production of aerobic energy, which is essential to normal brain development. Newborns are routinelly tested for blood concentration of Phe. The diet with low-phenylalanine diet. Downloaded from www.pharmacy123.blogfa.com
Catabolism of branched amino acids valine isoleucine leucine a-ketoglutarate glutamate (transamination) a-ketoisovalerate a-keto-b-methylbutyrate a-ketoisokaproate NAD+ oxidative decarboxylation Dehydrogenase of a-keto acids* CO2 NADH + H+ isovaleryl CoA a-methylbutyryl CoA isobutyryl CoA Dehydrogenation etc., similar to fatty acid b-oxidation acetyl CoA acetyl CoA propionyl CoA + + Downloaded from www.pharmacy123.blogfa.com propionyl CoA acetoacetate
Branched-chain aminoaciduria Disease also called Maple Syrup Urine Disease(MSUD) (because of the characteristic odor of the urine in affected individuals). Deficiency in an enzyme, branched-chain α-keto acid dehydrogenase leads to an accumulation of three branched-chain amino acids and their corresponding branched-chain α-keto acids which are excreted in the urine. There is only one dehydrogenase enzyme for all three amino acids. Mental retardation in these cases is extensive. Downloaded from www.pharmacy123.blogfa.com
Histidine Metabolism: Histamine Formation Histidine decarboxylase Histidine CO2 Histamine • Histamine: • Synthesized in and released by mast cells • Mediator of allergic response: vasodilation, bronchoconstriction Downloaded from www.pharmacy123.blogfa.com
Tryptophan catabolism • Tryptophan has complex catabolic pathway: • the indol ring is ketogenic • the side chain forms the glucogenic products • Kynurenate and xanthurenate are excrete in the urine. Downloaded from www.pharmacy123.blogfa.com
Enzymes which metabolised amino acides containe vitamines as cofactors THIAMINE B1 (thiamine diphosphate) oxidative decarboxylation of a-ketoacids RIBOFLAVIN B2 (flavin mononucleotide FMN, flavin adenine dinucleotide FAD) oxidses ofa-aminoacids NIACIN B3 – nicotinic acid (nikotinamide adenine dinucleotide NAD+ nikotinamide adenine dinukleotide phosphate NADP+) dehydrogenases, reductase PYRIDOXIN B6 (pyridoxalphosphate) transamination reaction and decarboxylation FOLIC ACID (tetrahydropholate) Meny enzymes of amino acid metabolism Downloaded from www.pharmacy123.blogfa.com
Helpful website http://themedicalbiochemistrypage.org/amino-acid-metabolism.html Downloaded from www.pharmacy123.blogfa.com