Download

1 / 27
290 likes | 757 Vues
Amino Acid Metabolism . Lecture 17 Modified from internet sources, books and journals. Introduction . All tissues have some capability for: synthesis of the non-essential amino acids

E N D
Amino Acid Metabolism Lecture 17 Modified from internet sources, books and journals
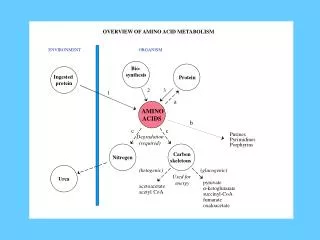
Introduction • All tissues have some capability for: • synthesis of the non-essential amino acids • conversion of non-amino acid carbon skeletons into amino acids and other derivatives that contain nitrogen • liver the major site of nitrogen metabolism in the body • times of dietary surplus the potentially toxic nitrogen of amino acids is eliminated via transaminations, deamination, and urea formation • carbon skeletons are generally conserved as: • carbohydrate, via gluconeogenesis • fatty acid via fatty acid synthesis pathways
continued • In this respect amino acids fall into three categories: • Glucogenic • Ketogenic • Glucogenic and ketogenic • Glucogenic amino acids those that give rise to a net production of pyruvate or TCA cycle intermediates (α-ketoglutarate or oxaloacetate) precursors to glucose via gluconeogenesis • All amino acids except lysine and leucine are at least partly glucogenic • Lysine and leucine the only amino acids that are solely ketogenic (giving rise only to acetylCoA or acetoacetylCoA)
continued • small group of amino acids (comprised of isoleucine, phenylalanine, threonine, tryptophan, and tyrosine) give rise to both glucose and fatty acid precursors are thus characterized as being glucogenic and ketogenic • amino acids have a third possible fate: during times of starvation the reduced carbon skeleton is used for energy production, with the result that it is oxidized to CO2 and H2O
Essential vs. Nonessential Amino Acids • Nonessential • Alanine, Asparagine, Aspartate, Cysteine, • Glutamate, Glutamine, Glycine, Proline, Serine, Tyrosine • Essential • Arginine*, Histidine, Isoleucine, Leucine, Lysine, Methionine*, Phenylalanine*, Threonine, Tyrptophan, Valine
continued • The amino acids arginine, methionine and phenylalanine considered essential for reasons not directly related to lack of synthesis • Arginine synthesized by mammalian cells but at a rate that is insufficient to meet the growth needs of the body and the majority that is synthesized is cleaved to form urea • Methionine required in large amounts to produce cysteine if the latter amino acid is not adequately supplied in the diet • phenyalanine needed in large amounts to form tyrosine if the latter is not adequately supplied in the diet
Alanine and the Glucose-Alanine Cycle • alanine second only to glutamine in prominence as a circulating amino acid • serves a unique role in the transfer of nitrogen from peripheral tissue to the liver • Alanine transferred to the circulation by many tissues, but mainly by muscle • In muscle alanine is formed from pyruvate • Liver accumulates plasma alanine, reverses the transamination that occurs in muscle, and proportionately increases urea production • When alanine transfer from muscle to liver coupled with glucose transport from liver back to muscle = the glucose-alanine cycle
continued • key feature of the cycle is that in 1 molecule alanine, peripheral tissue exports pyruvate and ammonia (which are potentially rate-limiting for metabolism) to the liver, where the carbon skeleton is recycled and most nitrogen eliminated • 2 main pathways to production of muscle alanine: • directly from protein degradation • via the transamination of pyruvate by alanine transaminase, ALT (also referred to as serum glutamate-pyruvate transaminase, SGPT)
continued • glucose-alanine cycle used primarily as a mechanism for skeletal muscle to eliminate nitrogen • Glucose oxidation produces pyruvate which can undergo transamination to alanine catalyzed by alanine transaminase, ALT • during periods of fasting skeletal muscle protein is degraded for the energy value of the amino acid carbons and alanine is a major amino acid in protein • alanine then enters the blood stream and is transported to the liver. in liver alanine is converted back to pyruvate which is then a source of carbon atoms for gluconeogenesis
Aspartate/Asparagine and Glutamate/Glutamine Biosynthesis • Glutamate is synthesized by the reductive amination of α-ketoglutarate catalyzed by glutamate dehydrogenase • glutamate arises by aminotransferase reactions, with the amino nitrogen being donated by a number of different amino acids glutamate is a general collector of amino nitrogen
continued • Aspartate is formed in a transamination reaction catalyzed by aspartate transaminase, AST • also formed by deamination of asparagine catalyzed by asparaginase
Amino Acid CatabolismGlutamine/Glutamate and Asparagine/Aspartate Catabolism • Glutaminase an important kidney tubule enzyme involved in converting glutamine (from liver and from other tissue) to glutamate and NH4+ • the NH4+ being excreted in the urine • Glutaminase activity in many other tissues as well, although its activity is not nearly as prominent as in the kidney
continued • Glutamate and aspartate important in collecting and eliminating amino nitrogen via glutamine synthetase and the urea cycle
Alanine Catabolism • Alanine important in intertissue nitrogen transport as part of the glucose-alanine cycle
Cysteine Catabolism • simplest, but least important pathway catalyzed by a liver desulfurase and produces hydrogen sulfide (H2S) and pyruvate • major catabolic pathway in animals via cysteine dioxygenase that oxidizes the cysteine sulfhydryl to sulfinate (producing the intermediate cysteinesulfinate) • Cysteinesulfinate serves as a biosynthetic intermediate e.g. precursor for the formation of 3'-phosphoadenosine-5'-phosphosulfate, (PAPS) • PAPS used for the transfer of sulfate to biological molecules such as the sugars of the glycosphingolipids
(other than protein), the most important product of cysteine metabolism is the bile salt precursor taurine • Taurine used to form the bile acid conjugates taurocholate and taurochenodeoxycholate
Valine, Leucine and Isoleucine Catabolism • catabolism of all three compounds initiates in muscle and yields NADH and FADH2 which can be utilized for ATP generation • principal product from valine propionylCoA, the glucogenic precursor of succinyl-CoA • Isoleucine catabolism terminates with production of acetylCoA and propionylCoA • Leucine gives rise to acetylCoA and acetoacetylCoA
Number of genetic diseases associated with faulty catabolism of aminoacids • most common defect in the branched-chain α-keto acid dehydrogenase • there is only one dehydrogenase enzyme for all three amino acids, all three α-keto acids accumulate and are excreted in the urine • disease = Maple syrup urine disease (characteristic odor of the urine in afflicted individuals) • Mental retardation in these cases is extensive • since these are essential amino acids they cannot be heavily restricted in the diet; life of afflicted individuals is short and development is abnormal • main neurological problems are due to poor formation of myelin in the CNS
Phenylalanine and Tyrosine Catabolism • Phenylalanine: • incorporation into polypeptide chains, and production of tyrosine • phenylalanine catabolism always follows the pathway of tyrosine catabolism • main pathway for tyrosine degradation involves conversion to fumarate and acetoacetate • Tyrosine equally important for protein biosynthesis as well as an intermediate in the biosynthesis of several physiologically important metabolites e.g. dopamine, norepinephrine and epinephrine
continued • As in phenylketonuria (deficiency of phenylalanine hydroxylase, PAH) deficiency of tyrosine aminotransferase (TAT) leads to hypertyrosinemia and the urinary excretion of tyrosine and the catabolic intermediates between phenylalanine and tyrosine • adverse neurological symptoms • The first inborn error in metabolism ever recognized = alkaptonuria was demonstrated to be the result of a defect in phenylalanine and tyrosine catabolism