Methods
Estimating the fraction of non-coding RNAs in mammalian transcriptomes Yurong Xin, Giulio Quarta, Hin Hark Gan, and Tamar Schlick Department of Chemistry and Courant Institute of Mathematical Sciences New York University, New York, NY, 10012. Introduction

Methods
E N D
Presentation Transcript
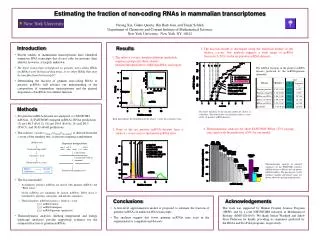
Estimating the fraction of non-coding RNAs in mammalian transcriptomes Yurong Xin, Giulio Quarta, Hin Hark Gan, and Tamar Schlick Department of Chemistry and Courant Institute of Mathematical Sciences New York University, New York, NY, 10012 • Introduction • Recent studies of mammalian transcriptomes have identified numerous RNA transcripts that do not code for proteins; their identity, however, is largely unknown. • Do these transcripts correspond to genuine non-coding RNAs (ncRNAs) with biological functions, or to other RNAs that may be non-functional transcripts? • Determining the fraction of genuine non-coding RNAs in putative ncRNAs will advance our understanding of the composition of mammalian transcriptomes and the general importance of ncRNAs for cellular function. Results • The fraction model is developed using the statistical feature of the relative z-score. Our analysis suggests a wide range of ncRNA fractions (5–50%) in the six putative ncRNA datasets. • The relative z-score classifies different nucleotide sequence groups into three clusters: genome/intergene/intron, mRNA/ncRNA, and repeat. The ncRNA fractions in the putative ncRNA datasets predicted by the ncRNA/genome submodel. • Methods • Six putative ncRNA datasets are analyzed: (1) FANTOM3 ncRNAs, (2) FANTOM3 stringent ncRNAs, RNAz predictions (3) set1.P0.5 (P>0.5), (4) set1.P0.9 (P>0.9), (5) set2.P0.5 (P>0.5), and (6) EvoFold predictions. • The relative z-score (zrelative=zbioseq/zdi-random) is derived from the z-score of the monkey test; it assesses sequence randomness. • The fraction model • Assumption: putative ncRNAs are mixed with genuine ncRNAs and “RNA noise”. • Novel ncRNAs are simulated by known ncRNAs; RNA noise is simulated by genomic, intergenic, and intronic sequences. • Three functions: ncRNA fraction vs. relative z-score. • f1(z): ncRNA/intron • f2(z): ncRNA/intergene • f3(z): ncRNA/genome (preferred). • Thermodynamic analyses (melting temperature and energy landscape analyses) provide supporting evidence for the estimated fraction of genuine ncRNAs. The three functions of the fraction model are shown in solid lines. The dashed lines (a-f) indicate relative z-score of the six putative ncRNA datasets. Each line indicates the distribution of the relative z-score for a sequence class. • Thermodynamic analyses for short FANTOM3 RNAs (23% passing rate) agree with the prediction (18%) by our model. • None of the six putative ncRNA datasets have a relative z-score close to the known ncRNA class. Monkey test Sequence manipulation seq-2 seq-n seq-1 … Count missing motifs Concatenate Calculate z-score Cut into 2097152-nt segments Obtain relative z-score Thermodynamic analyses of selected sequences of the FANTOM3 putative ncRNA dataset (<400 nt) and ten known ncRNA families. The passing rate, tested sequence number and dataset name are shown above the passing frequency bar. Higher randomness Smaller z-score Submit to the monkey test • Conclusions • A first-level approximation model is proposed to estimate the fraction of genuine ncRNAs in unknown RNA transcripts. • The analyses suggest that fewer genuine ncRNAs may exist in the experimental or computational datasets. Acknowledgements The work was supported by Human Frontier Science Program (HFSP) and by a joint NSF/NIGMS initiative in Mathematical Biology (DMS-0201160). We thank Stefan Washietl and Jakob Skou Pedersen for kindly providing us sequences predicted by the RNAz and EvoFold programs, respectively.