Download
1 / 15
160 likes | 458 Vues
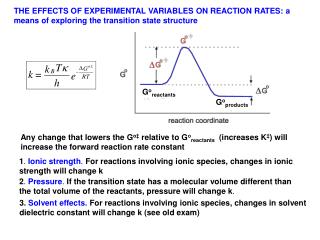
THE EFFECTS OF EXPERIMENTAL VARIABLES ON REACTION RATES: a means of exploring the transition state structure. G o reactants. G o products. Any change that lowers the G o‡ relative to G o reactants (increases K ‡ ) will increase the forward reaction rate constant .

E N D
THE EFFECTS OF EXPERIMENTAL VARIABLES ON REACTION RATES: a means of exploring the transition state structure Goreactants Goproducts Any change that lowers the Go‡ relative to Goreactants (increases K‡) will increase the forward reaction rate constant 1. Ionic strength. For reactions involving ionic species, changes in ionic strength will change k 2. Pressure. If the transition state has a molecular volume different than the total volume of the reactants, pressure will change k. 3. Solvent effects. For reactions involving ionic species, changes in solvent dielectric constant will change k (see old exam)
fast fast E + S ES ES‡ EP E + P ES ES‡ 2. Pressure dependence of an enzyme reaction rate The overall reaction rate will be effected by anything that changes the free energy of the activated complex ES‡ relative to the ES complex, that is, anything that changes the “equilibrium” Suppose that the molecular volume of the enzyme changes in forming the activated complex such that V(ES) ≠ V(ES‡) In this case, the relative free energy of the activated complex will depend on pressure
(assuming is pressure independent) (a familiar equation now applied to an activated complex) DG‡ is the free energy of activation at P atmospheres. The rate constant at pressure P is then Where , the rate constant at P =1atm
0 ln(k) – ln(ko) P-1 A volume change in forming the activated complex can be directly measured from the pressure dependence of the rate constant For pyruvate kinase, DV‡ = +30 cm3/mole. For reference, the molar volume of the enzyme is around 20,000 cm3/mole. Thus the volume change on activation is around 0.1%. The rate is reduced with pressure, because the volume change is positive. At the bottom of the Mariana trench (depth of 30,000 ft), P = 1000 atm. At this pressure k/ko = 0.27; The enzyme rate constant is reduced by about 70%. Has the enzyme for organisms that reside at the bottom evolved for a reduced volume of activation?
A+ + B- [AB]‡ → Products 3. Solvent effects on k (old final problem exam) Consider the elementary reaction in a solution of I = 0 Derive an equation for ln(k2/k1) where k2 and k1 are the rate constants for the reaction in solvents of dielectric constant e2 and e1, respectively. But the activated complex is uncharged, so unaffected by the dielectric constant
Both A and B are charged; the terms in brackets above are then just the Born free energies of transfer from a solvent of dielectric constant e1 to one of e2. Because Z2 is the same for both A and B
A+ + B- [AB]‡ → Products If e2 > e1, k2 < k1. Why? Because A and B are charged, their free energy is lower in a higher dielectric constant due to favorable interactions with the solvent dipoles. The activated complex is not effected, so the activation free energy increases.
REVIEW OF TOPICS: Chem 156 Spring 2005 I. FUNDAMENTALS OF THERMODYNAMICS (18%) • The second law and entropy; criterion of spontaneity in terms of entropy (DS 0, isolated system); statistical interpretation of entropy (S =RlnW). • Free energy as a criterion of spontaneity at constant T, P for changes involving work other than PV work (dG –dW’) 0). • The total chemical potential and general conditions for equilibrium at constant T,P • (i) physical equilibria: for all phases or regions • (ii) Chemical equilibria:
Chemical potentials in solution (i) Standard states for solvents and solutes (Raoult’s and Henry’s laws) (ii) Physical interpretation of mo and g in terms of solvent-solute and solute-solute interactions, respectively (non-electrolyte solutions) II. THERMODYNAMICS OF SOLUTIONS (15%) Principles First applications of general equilibrium conditions and equations for m 1. Chemical equilibria in non-ideal solutions DG = DGo + RTlnQ • 2. Free energy coupling in Biochemistry (Bioenergetics): ATP-dependent Ca++ transport in the absence of a membrane potential; energy coupling in solution reactions (DGsource + DG process = 0) • 3. Sedimentation equilibrium ; density • gradients; equilibrium in the gravitational field;
III. ELECTROSTATIC EFFECTS IN BIOCHEMISTRY (25%) • Basic electrostatics; definition of electrostatic potential; electrostatic free energy DG = qDF = ZFDF • Electrochemical equilibria • (i) distribution of ions in potential gradients • (ii) Origin of the transmembrane potential (diffusion potential) • (iii) Application: effect of transmembrane potential on membrane equilibria; the Chemiosmotic theory of oxidative phosphorylation (other examples in problem sets an on exam I) • 3. Ion-solvent interactions: the Born Model • dielectric constant: determines strength of ion-solvent interaction; origin is dipole moment and polarizibility of solvent • (ii) Born model for the free energy, enthalpy and entropy of ion solvation
3.Ion-solvent interactions: the Born Model (iv) Applications: membrane permeability, action of ionophores and the contribution to protein stability • 4.The origin of electrostatic potentials around proteins and near membranes • The Debye-Huckel model for uniformly charged spherical proteins: F(r); the “ion cloud”; k and the spatial dependence of the potential; validity of the assumptions. • Applications: local concentrations of ions at the surface (F(a)) relative to the “bulk” solution (F() = 0); effect of surface potential on ionization of surface groups; contribution of surface potential to protein stability (effect of Z (pH), ionic strength). • The Gouy-Chapman model for charged membrane surfaces: F(x); k and the spatial dependence of the potential. • Applications: local concentration of ions at the membrane surface (same as for proteins)
IV. THE HYDROPHOBIC EFFECT (7%) • The solubility of alkanes and amphiphiles in water; the free energy of transfer (uow – uonp); principle of group contributions to the free energy of transfer (additivity of the free energies). • The temperature dependence of the free energy of transfer, and the definition of the hydrophobic effect( for transfer to water near 298K, DGo>0, DHo small, DSo <0, dominant, DCp > 0 (T dependence of DHo, DSo) ). • Molecular origin(stabilization of ordered structures in water, at least near room temperature) V. APPLICATION OF TOPICS I-IV TO UNDERSTANDING THE PRINCIPLES OF SELF-ASSEMBLING SYSTEMS IN BIOCHEMISTRY (15%) • Micelles(7%) • The phase separation model and the CMC; thermodynamic stability of the micelle (hydrophobic effect); group contributions to the stability. • Size and shape of the micelle; principle of opposing forces and the role of head-group repulsion; the phospholipid bilayer
Proteins (8%) • The Tanford calculation and protein stability; quantitative role of the hydrophobic effect, the configurational entropy and the H-bond. • Updates to the Tanford calculation: potentially more important role of the H-bond, but phenomena such as “cold denaturation” emphasize the importance of the hydrophobic effect; the configurational entropy of a folded protein is not 0! • Stability of an isolated a-helix; cooperativity in protein folding VI. CHEMICAL KINETICS (20%) • Empirical rate laws: experimental determination from initial rates and integrated rate laws • Elementary reactions and mechanisms; rate laws for elementary reactions (unimolecular, bimolecular, termolecular) • Rate laws for complex reactions; rate limiting steps, pre-equilibrium and steady-state approximations for obtaining rate laws
VI. CHEMICAL KINETICS (20%) • 4. Determining a rate law from a mechanism (see oxidation of NO, renaturing DNA, membrane transport and problem set 10 for examples) • 5. Temperature dependence of k; the empirical Arrhenius equation • 6. Transition state theory of the rate constant; free energy, enthalpy and entropy of activation; molecular interpretations • (i) Applications • Ionic strength dependence of k for ionic reactions • Pressure dependence of k • Solvent dependence of k (problem from old exam)
Study guide for final exam • The final will contain some (probably 4) multiple choice questions of 5 parts each covering: (1) general thermodynamics and solution thermodynamics (2) electrostatics; (3) the hydrophobic effect, micelle formation and proteins and (4) kinetics. See problem set 10 for examples. • In addition, there will be calculational problems (probably 7) of the kind on the previous 2 exams. See problem set 10 for examples of kinetics problems.