Download
1 / 51
590 likes | 1.04k Vues
We know that each irreducible rep is 1 dimensional but let’s assume that we do not know the dimensions of the irreducible reps.

E N D
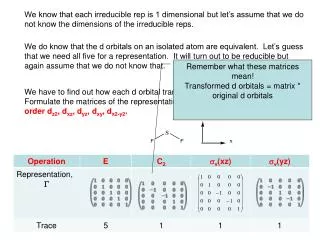
We know that each irreducible rep is 1 dimensional but let’s assume that we do not know the dimensions of the irreducible reps. We do know that the d orbitals on an isolated atom are equivalent. Let’s guess that we need all five for a representation. It will turn out to be reducible but again assume that we do not know that. Remember what these matrices mean! Transformed d orbitals = matrix * original d orbitals We have to find out how each d orbital transforms under the C2v operations. Formulate the matrices of the representation based on the d orbitals in this order dz2, dxz, dyz, dxy, dx2-y2.
As the distance between atoms decreases Atomic orbitals overlap Bonding takes place if: the orbital symmetry must be such that regions of the same sign overlap the energy of the orbitals must be similar the interatomic distance must be short enough but not too short If the total energy of the electrons in the molecular orbitals is less than in the atomic orbitals, the molecule is stable compared with the atoms
Antibonding Bonding More generally: Y = N[caY(1sa) ± cbY (1sb)] n A.O.’s n M.O.’s Combinations of two s orbitals (e.g. H2)
Electrons in antibonding orbitals increase the energy of the system. Electrons in bonding orbitals concentrate between the nuclei and hold the nuclei together (total energy is lowered)
Not s Both s and s* notation indicates symmetric with respect to rotation about internuclear axis s* s s*
p orbitals can combine in two ways, s and p. s (and s*) notation means no change of sign upon rotation p (and p*) notation means change of sign upon 180 rotation
Combination of s and p orbitals No overlap, no bonding Combination of two misaligned p orbitals in p mode.
d orbitals overlapping s type overlap: symmetric for any rotation No interaction – different symmetry p type overlap: antisymmetric for 180 rotation, d type overlap. Changes sign for rotation by 90
Is there a net interaction? NO NO YES
Relative energies of interacting orbitals must be similar Weak interaction Strong interaction
Molecular orbitals for diatomic molecules From H2 to Ne2 Electrons are placed in molecular orbitals following the same rules as for atomic orbitals: Fill from lowest to highest Maximum spin multiplicity Electrons have different quantum numbers including spin (+ ½, - ½)
O2 (2 x 8e) 1/2 (10 - 6) = 2 A double bond Or counting only valence electrons: 1/2 (8 - 4) = 2 Note subscripts g and u symmetric/antisymmetric upon i
Place labels g or u in this diagram s*u p*g pu sg
s*u sg g or u? p*g pu d*u dg
Orbital mixing In this diagram some of the MOs have the same symmetry. This allows for interaction to take place. Mixing can occur. Orbitals of sg symmetry can mix together to yield a more stable and a less stable combination. Likewise for su For the orbitals of sg symmetry.
Overall result: s orbital mixing Notice the change in ordering here caused by the mixing. Li N O Ne When two MO’s of the same symmetry mix the one with higher energy moves higher and the one with lower energy moves lower. This revised ordering occurs Li N, but not O Ne
The electronic configuratations O2 bond order 2 F2 bond order 1 N2 bond order 3 B2 bond order 1 C2 bond order 2 Be2 bond order ? Li2 bond order 1 Expect the shortest bond length for N2 (But remember the atomic radii decrease with F smallest.)
Filling bonding orbitals Filling antibonding orbitals Bond lengths in diatomic molecules
Paramagnetic due to mixing Neutral: C2pu2 pu2 (double bond) Dianion: C22- pu2 pu2sg2(triple bond) In Li2 through N2sg above pu. O2 through Ne2 pu above sg. -The s orbitals are more affected by effective nuclear charge than are the p. -Less mixing for O, F and Ne due to smaller orbitals and greater bond distance. Neutral: O2pu2 pu2 p*g1p*g1 (double bond, paramagnetic) Dianion: O22- pu2 pu2 p*g2p*g2 (single bond, diamagnetic)
Ionization is a transition between states • Initial State: Neutral (or anion) • Final State: Atom/Molecule/Anion after an electron is removed, plus the ejected electron • M → M+ + e- init = M ; final = M+ + e- • For direct photoionization, transition probability is always > 0
e- + Molecule+ hn + Molecule = EM+ - EM Ehn - Ee- = Ionization Energy IE = Difference in energy between states of M, M+
18 17 16 15 Ionization Energy (eV) What about molecules? σ* ↿ ⇂ H 1s H 1s ↿⇂ σ
Transitions between molecular potential energy surfaces Excited State During an electronic transition the complex absorbs energy electrons change orbital the complex changes energy state molecular rotations lower energy microwave radiation electron transitions higher energy visible and UV radiation Ground State Timescale : ≈10-15 sec Timescale of geometry changes (vibrations): ≈10-12 sec As a result, observe vertical (Franck-Condon) transitions In other words, we assume that we only have to consider the electronic portion of the ground- and excited-statewavefunctions to understand these transitions: Born-Oppenheimer approximation molecular vibrations medium energy IR radiation
H2+ 18 17 16 15 Ionization Energy (eV) H2 0 1 0 2 r (Å) Potential Energy Surface Description of the Ionization of Dihydrogen Much more on this next time!!
20 19 18 17 16 15 Ionization Energy (eV) Consider Dinitrogen First ion state (X) = 2g+ 2u Second ion state (A) = 2u 1g Third ion state (B) = 2u+ 2p 2p 2g+ 1u 2s 2s 1u+ 1g+ :N≡N: Ground state (X) = 1g+
2 + S u 2 + P u 2 + S g Potential Well Description 2u 1g 2p 2p 2g+ 1u 2s 2s 1u+ 1g+ 1 S N 2 :N≡N: g Ground state (X) = 1g+
Models to describe molecular electronic structure MO Theory compared to Valence Bond Theory
CH CH 2p 4 4 sp3 2s 24 24 22 22 20 20 18 18 16 16 14 14 12 12 Ionization Energy (eV) Ionization Energy (eV) Consider methane. VSEPR gives 4 sp3 hybrid orbitals. Photoelectron Spectroscopy So why are there two valence ionizations separated by almost 10 eV?
Use of reducible representations in M.O. theory Consider transformation properties of vectors aligned with the 4 C-H bonds.
Apply Reduction Formula: C-H = A1 + T2 This is what the bonding MOs orbitals should be. http://www.mpip-mainz.mpg.de/~gelessus/group.html
CH CH 4 4 24 24 22 22 20 20 18 18 16 16 14 14 12 12 Ionization Energy (eV) Ionization Energy (eV) LCAO Description of Methane, Td 2p (t2) t2 (1, 2, 3) a1 (1) 2s (a1) C CH4 H4 Obtained using the carbon AOs as basis objects. Obtained using the hydrogen AOs as basis objects. Orbitals of same symmetry combine and bond.
Now back to our ordering of the MOs of diatomics. Orbital Energy Orbital Energy N2 O2 sg (2p) p*u (2p) pu (2p) sg (2p) pu (2p) Very involved in bonding (vibrational fine structure) s*u (2s) s*u (2s) (Energy required to remove electron, lower energy for higher orbitals)
Very Simple Molecular Orbital Theory A molecular orbital, f, is expressed as a linear combination of atomic orbitals, holding two electrons. The multi-electron wavefunction and the multi-electron Hamiltonian are Where hi is the energy operator for electron i and involves only electron i
Seek F such that Divide by F(1,2,3…) recognizing that hi works only on electron i. Since each term in the summation depends on the coordinates of a different electron then each term must equal a constant.
Now our attention is on the equation that only involves one electron. Recall the expansion of a molecular orbital in terms of the atomic orbitals. Multiply equation at top by one of the atomic orbitals, uk k = 1,2,…#AOs, and integrate. Now use the expansion of the MO in the AOs. These integrals are fixed numerical values. Define The al, l = 1, 2, 3…#AOs are unknowns. These values are known numerical quantities. There are #AO such equations. Secular equations.
For k = 1 to AO These are the secular equations. The number of such equations is equal to the number of atomic orbitals, AO. The number of equations is equal to the number of AOs equations. The number of unknowns, the al, is also equal to the number of AOs. For there to be a nontrivial solution (all al not equal to zero) to the set of secular equations then the determinant below must equal zero
Drastic assumptions can now be made. We will use the simple Huckel approximations. hi,i = a, if orbital i is on a carbon atom. Si,i = 1, normalized atomic orbitals, hi,j = b, if atom i bonded to atom j, zero otherwise Sij = 0 if I is not equal to j • Expand the secular determinant into a polynomial of degree AO in e, the energy. • Obtain the allowed values of e by finding the roots of the polynomial. • Choose one particular value of e, substitute into the secular equations. • Obtain the coefficients of the atomic orbitals within the molecular orbital.
Example The allyl pi system. The secular equations: (a-e)a1 + b a2 + 0 a3 = 0 ba1 + (a-e) a2 + b a3 = 0 0 a1 +b a2 + (a-e) a3 = 0 Simplify by dividing every element by b and setting (a-e)/b = x
For x = -sqrt(2) e = a + sqrt(2) b For x = 0; e = a Substitute into the secular equations normalized
Verify that h f = e f
Perturbation Theory H0 is the Hamiltonian of for a known system for which we have the solutions: the energies, e0, and the wavefunctions, f0. H0f0 = e0f0 We now change the system slightly (change a C into a N, create a bond between two atoms). The Hamiltonian will be changed slightly. For the changed system H = H0 + H1 H1 is the change to the Hamiltonian. We want to find out what happens to the molecular orbital energies and to the MOs.
Changes (approximate) Energy Zero order (no correction): ei0 First Order correction: Wave functions corrections to f0i Zero order (no correction): f0i First order correction:
Example Pi system only: Perturbed system: allyl system Unperturbed system: ethylene + methyl radical
Mixes in anti-bonding Mixes in bonding Mixes in anti-bonding Mixes in bonding