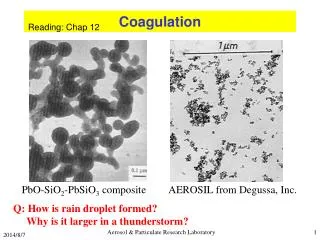
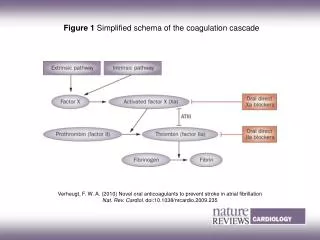
Coagulation Cascade
Coagulation Cascade. Amplification. Initiation. Introduction. Congenital bleeding disorder caused by low levels of specific coagulation factors Hemophilia A: 85%, factor VIII deficiency Third most common X-linked disorder Hemophilia B: 10%-15%, factor IX deficiency. Genetics.
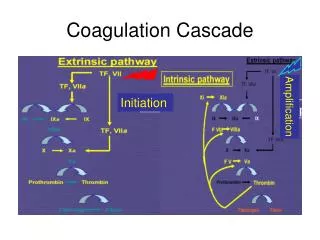

Coagulation Cascade
E N D
Presentation Transcript
Coagulation Cascade Amplification Initiation
Introduction • Congenital bleeding disorder caused by low levels of specific coagulation factors • Hemophilia A: 85%, factor VIII deficiency Third most common X-linked disorder • Hemophilia B: 10%-15%, factor IX deficiency
Genetics • X-linked recessive disorder • FVIII gene is on Xq28; most common (45%) defect is inversion and translocation of exons 1-22 away from 23-26, others: point mutation • 1/10,000 live male births • 女性也可能為symptomatic carriers (ex. extreme “Lyonisation” of the normal X chromosome) • 約1/3為mutation
Classifications 依血液中凝血因子的剩餘含量而定 • Severe: < 1 unit/dL (1% activity) • Moderate: 1%-5% • Mild: > 5% Definition • 1 unit = the amount found in 1ml of normal pool plasma • 100% activity = the actvity found in 1ml of normal pool plasma
Bleeding Manifestations in Hemophilia Sites of bleeding • Serious Joints (hemarthrosis), muscle/soft tissue, mouth/gum/nose, hematria • Life-threatening CNS, GI, neck/throat, severe trauma • Incidence of different sites of bleeding Hemarthrosis: 70-80% Muscle/soft tissue: 5-10% CNS: <5%
Clinical Manifestations • 嚴重者,出生時就可能出現subgaleal hematoma and/or ICH〈應特別留意嬰兒室頭圍迅速增大的新生兒〉 • 症狀可能多在開始爬及走路時才出現 • The hallmark of hemophilic bleeding:自發性的關節出血 (hemarthrosis) and intramuscular hematoma • 1-2 % ICH
其他出血的警訊 • 腦出血:infants with “meningitis” • 後腹腔出血:嚴重腹痛,可能誤為盲腸炎 • 頸部、咽喉出血:喉痛、吞嚥困難,可能阻塞呼吸道 • Toddler: ankle most, than knee • Child: knee most, than ankle
Diagnosis and Laboratory Tests • Family history • Normal platelet count, bleeding time, PT • Prolonged PTT • Specific factor assays • Genetic testing • Prenatal diagnosis
Management • History of hemophilia treatment
治療 • A型: FFP,cryoprecipitate 或 factor VIII concentrate (dose: desired rise level % × BW × 0.5) • B型:FFP或factor IX concentrate (dose: desired rise level % × BW × 1.2~1.5) • Factor concentrate: plasma-derived or recombinant product • DDAVP for mild or moderate form • Antifibrinolytic therapy
On-demand Therapy • 一有症狀出現就及早注射(即補充)凝血因子 • 劑量及次數視體重、病情輕重、出血程度及出血部位而定
Recommeded Plasma Factor Level and Duration of Administration
Prophylactic Therapy • 原理:將重度缺乏者的凝血因子提升至 >1% • 有效減少自發性出血 • 避免關節出血及進一步關節病變 • 瑞典最早開始, A型血友病自1958年起,B型血友病自1972年起
Prophylaxis • Initial observation: persons with moderate hemophilia (1-5% FVIII) have decreased joint disease • Hypothesis: converting a person from severe hemophilia to moderate with prophylaxis would decrease incidence of joint disease • Goat: to raise FVIII above 1% was commenced in Malmo, Sweden in 1958 • Lovqvist, et al: J Intern Med 1997
When to Start: The Swedish Experience Conclusion: prophylaxis should be started in the first years of life, before age 3 Astermark et al: Br J Hematol 1999
一般照護 • 預防注射: 最小的針頭接種 • 不可接受其他肌肉注射 • 勿吃含有阿斯匹靈的止痛藥, 但Panadol, Ponstan, Codeine可以服用 • 牙齒的保護
長期的關節病痛問題 • Target joints: 膝關節、踝關節及肘關節 • 惡性循環下,發生慢性關節炎 • Arthropathy: most significant chronic morbidity • 預防是最佳的處理方法 • Arthroscopic synovectomy • Joint replacement
輸血引發的感染 • 經加熱等特殊程序處理血液製品後,1985年以後出生的血友病患者,至今並無因注射凝血因子而感染愛滋病的報告 • HBV, HCV infection (vaccination using is recommended) • Recombinant product可完全免除這種潛在的危險
凝血因子抗體 • 14-25% of severe hemophilia A, very rare in hemophilia B • 可能與基因有關 • 輕者可能需要大量而頻繁的注射才可能止血 • 嚴重的就可能對注射凝血因子無效,必須使用其他凝血因子 (by-pass)
What are inhibitors ? • Antibodies directed against coagulation factors Alloantibody in patients with hemophilia A or B Autoantibody in people without hemophilia • Incidence Antifactor VIII inhibitors in hemophilia A: 25% Antifactor IX inhibitors in hemophilia B: 1-3% Antifactor VIII autoantibody inhibitors: 1/106/year • Usually result in loss of coagulation factor function
Factor VIII inhibitors • The most common inhibitor • Polyclonal IgG antibodies, esp IgG4 • Bleeding is more severe in autoantibody patients than in hemophilia A inhibitor patients
EtiologyDefinite Factors Involved • FVIII gene mutation No FVIII protein means high risk • Adjuvants in FVIII products • Race Higher in African-Americans • HLA status • Immune modifiers IL10 polymorphism
Classification of Inhibitors Definitions • “High” responders IgG inhibitors of titer > 5 Bethesda Units (BU) Inability to overwhelm with native factor • “Low” responders < 5 BU Transient Less likely to have anamnestic responses Amenable to treatment by overwhelming inhibitor with native factor VIII or XI
Treatment Options for High-responder Inhibitors Bypassing agents • Low-purity, plasma-derived concentrates Prothrombin complex concentrates Activated prothrombin complex concentrates Recombinant VIIa • Emergency treatments Recombinant VIIa Plasmapheresis Porcine VIII and new recombinant procine VIII
Treatment Options for High-responder Inhibitors • Immune tolerance induction • Rituximab Anti-CD20 chimeric antibody reliably depletes peripheral B cells Several reports of success in acquired hemophilia (an autoimmune disorder) NHLBI-sponsored clinical trial through Transfusion Medicine/Hemostasis research network to begin May 2006 • Fox et al, Hemophilia 2006
von Willebrand Disease • Disorder first described by Erik von Willebrand in 1925 in persons living off the coast of Finland • Marked heterogeneity in phenotype, autosomal Dominant or Recessive Inheritance • Deletion in chromosome 12 is most common • Overall prevalence 1:100 to 1: 500 • Incidence equal among Man and Women (chromosome 12)
vWF Genetics • http://www.vwf.group.shef.ac.uk/pictures.html Location of Gene: chromosome 12 (p13.3)
vWF Protein • A1: binds to Gp IB alpha • A3: Collagen binding Domain • D’/D3: Interacts with Factor VIII • C2: Interacts with GpIIb/IIIa • http://www.vwf.group.shef.ac.uk/pictures.html
vWF Protein • A1: type 2B and 2M • A2: type 2A, cleavage site for ADAMTS 13 • D’/D3: type 2N • http://www.vwf.group.shef.ac.uk/pictures.html
vWD Tests: Initial Work-up • Quantitative • Factor VIII level • vWF Antigen level • vWF Multimers • Qualitative • Ristocetin Cofactor Assay: studies function of Vwf/Platelet interaction
Official Abbreviated Terms as Designated by ISTH ISTH: International Society on Thrombosis and Hemostasis
Differential Diagnosis Hemophilia A & von Willebrand Disease
Management • Education • Cryoprocipitate (dose: desired rise level % × BW × 0.75) • DDAVP for type 1 • Amicar (antifibrinolytic agent) for mucosal bleeds • Humate-P (factor 8 and vWF) for surgery, trauma • Platelet for pseudo-vWD • Recombinant factor 7a, correct underlying disorder (hypothyroidism) for acquired vWD
Platelet Anatomy • Disc-shaped, anuclear fragment • Size: 1.5 μm • Normal maturation time 4-5 days • Circulating life span 9-10 days
Platelet Anatomy • Peripheral zone Plasma membrane Open canalicular system Extension of the plasma membrane Forms interconnecting network, greatly increases the surface area Membrane proteins: receptors for agonists and adhesive glycoproteins, signal transduction molecules IIb-IIIa: fibrinogen, vWF, fibronectin Ib-IIa: collagen Ib-IX-V: insoluble Vwf VI: collagen
Platelet Anatomy • Submembranous zone Contractile protein system: regulates shape and carry out events such as secretion of granules and retraction of clots • Organelle zone Platelet specific storage granules Dense bodies: serotonin, ADP, ATP, Ca α granules: platelet factor 4, thromboglobulin, PDGF, vWF Lysosomes, peroxisomes