Download

1 / 81
1.19k likes | 3.68k Vues
The Organic Chemistry of Drug Design and Drug Action. Chapter 3 Receptors. 1878 Langley Study of antagonistic action of alkaloids on cat salivary flow suggests the compounds interacted with some substance in the nerve endings 1897 Ehrlich

E N D
The Organic Chemistry of Drug Design and Drug Action Chapter 3 Receptors
1878 Langley Study of antagonistic action of alkaloids on cat salivary flow suggests the compounds interacted with some substance in the nerve endings 1897 Ehrlich Side chain theory - Cells have side chains that contain groups that bind to toxins - termed receptors 1906 Langley Studying antagonistic effects of curare on nicotine stimulation of skeletal muscle Concluded receptive substance that received stimulus, and by transmitting it, caused muscle contraction
Two fundamental characteristics of a receptor: • Recognition capacity - binding • Amplification - initiation of response
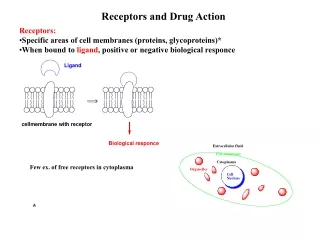
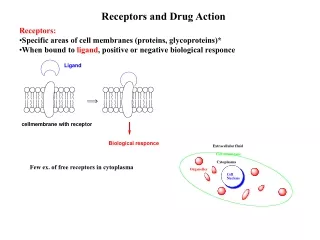
Integral proteins embedded in phospholipid bilayer of membranes Figure 2.14
Drug-Receptor InteractionsPharmacodynamics (3.1) Driving force for drug-receptor interaction - low energy state of drug-receptor complex (binding energy) Kd - measure of affinity to receptor (a dissociation constant)
Forces Involved in Drug-Receptor Complex Molecular surfaces must be close and complementary G°= -RTlnKeq (3.2) Decrease in G°of ~ 5.5 kcal/mol changes binding equilibrium from 1% in drug-receptor complex to 99% in drug-receptor complex Forces in drug-receptor complex generally weak and noncovalent (reversible)
Ionic Interaction Basic groups, e.g., His, Lys, Arg (cationic) Acidic groups, e.g., Asp, Glu (anionic) Figure 3.1 G°≈ -5 kcal/mol
Ion-Dipole and Dipole-Dipole Interactions G°≈ -1 to -7 kcal/mol Figure 3.2
Hydrogen Bonding Type of dipole-dipole interaction between H on X-H (X is an electronegative atom) and N, O, or F G°≈ -3 to -5 kcal/mol Figure 3.3
3.5 a-helix
-sheet 3.6
DNA 3.7
Charge-Transfer Complexes(molecular dipole-dipole interaction) chlorothalonil- fungicide acceptor donor G°≈ -1 to -7 kcal/mol Figure 3.5
Hydrophobic “Interactions” Increase in entropy of H2O molecules decreases free energy. Therefore the complex is stabilized. Figure 3.6
Hydrophobic Interaction butamben - topical anesthetic Figure 3.7 G°≈ -0.7 kcal/mol per CH2/CH2 interaction
Van der Waals Forces As molecules approach, temporary dipoles in one molecule induce opposite dipoles in another; therefore, producing an intermolecular attraction G°≈ -0.5 kcal/mol per CH2/CH2 interaction
Figure 3.8 Dibucaine - local anesthetic
Dose-Response Curve % Muscle Contraction Use any measure of response (LD50, ED50, etc.) Figure 3.9 Means of measuring drug-receptor interactions
Full Agonist Figure 3.10
Antagonists Competitive Antagonist Noncompetitive Antagonist Different binding sites Figure 3.11
Partial Agonist low [neurotransmitter] added agonist effect high [neurotransmitter] added antagonist effect Figure 3.12
Inverse Agonists full inverse agonist Addition of an agonist or antagonist to an inverse agonist (a, b, c are increasing concentrations of agonist added) partial inverse agonist Figure 3.13
To effect a certain response of a receptor, design an agonist • To block a particular response of a natural ligand of a receptor, design an antagonist • To produce the opposite effect of the natural ligand, design an inverse agonist
Two stages of drug-receptor interactions: 1) complexation with receptor 2) initiation of response (Stephenson) (Ariëns) efficacy intrinsic activity affinity 5 different drugs = 1 full agonist < 1 partial agonists All are full agonists Figure 3.15
Affinity and efficacy are uncoupled: a compound can have great affinity but poor efficacy (and vice versa). A compound can be an agonist for one receptor and an antagonist or inverse agonist for another receptor. A full or partial agonist displays positive efficacy. An antagonist displays zero efficacy. A full or partial inverse agonist displays negative efficacy.
Table 3.1 Agonists - often structural similarity Antagonists - little structural similarity
How can agonists and antagonists bind to same site and one show response, other not? Figure 3.14 agonist antagonist enantiomer • All naturally-occurring chemicals in the body are agonists • Most xenobiotics are antagonists • Drugs that bind to multiple receptors side effects
Intensity of pharmacological effect is directly proportional to number of receptors occupied Does not rationalize how two drugs can occupy the same receptor and act differently Drug-Receptor Theories Occupancy Theory (1926)
Rate Theory (1961) Activation of receptors is proportional to the total number of encounters of a drug with its receptor per unit time. Does not rationalize why different types of compounds exhibit the characteristics they do.
Induced Fit Theory (1958) • Agonist induces conformational change - response • Antagonist does not induce conformational change - no response • Partial agonist induces partial conformational change - partial response Figure 3.16
Activation-Aggregation TheoryMonad, Wyman, Changeux (1965) Karlin (1967) Receptor is always in a state of dynamic equilibrium between activated form (Ro) and inactive form (To). Ro To biological response no biological response Agonists shift equilibrium to Ro Antagonists shift equilibrium to To Partial agonists bind to both Ro and To Binding sites in Ro and To may be different, accounting for structural differences in agonists vs. antagonists
Two-state (Multi-state) Receptor Model R and R* are in equilibrium (equilibrium constant L), which defines the basal activity of the receptor. Full agonists bind only to R* Partial agonists bind preferentially to R* Full inverse agonists bind only to R Partial inverse agonists bind preferentially to R Antagonists have equal affinities for both R and R* (no effect on basal activity) In the multi-state model there is more than one R state to account for variable agonist and inverse agonist behavior for the same receptor type. Figure 3.17
Drug and Receptor ChiralityDrug-Receptor Complexes Receptors are chiral (all L-amino acids) Racemic mixture forms two diastereomeric complexes [Drug]R + [Drug]S + [Receptor]S [Drug]R [Receptor]S+ [Drug]S [Receptor]S Have different energies and stabilities
Topographical and Stereochemical ConsiderationsSpatial arrangement of atoms Common structural feature of antihistamines (antagonists of H1 receptor) Pharmacophore - parts of the drug that interact with the receptor and cause a response Figure 3.18 CH-O, N-, CH- 2 or 3 carbons
Chiral antihistamine Kd for enantiomers are different - two diastereomers are formed (S)-(+)-isomer 200x more potent than (R)-(-)- More potent isomer - Less potent isomer - eutomer distomer Ratio of potencies of enantiomers - High eudismic ratio when antagonist has stereogenic center in pharmacophore eudismic ratio
Distomer is really an impurity (“isomeric ballast”) May contribute to side effects and/or toxicity (S)-(-)-thalidomide teratogen 3.13 (R)-(+)-thalidomide sedative/hypnotic
Enantiomers can have different activities 3.17 dextropropoxyphene (Darvon®) analgesic 3.18 levopropoxyphene (Novrad®) antitussive (anticough)
Enantiomers can have opposite activities barbiturate S-(+)- convulsive R-(-)- narcotic (actually inverse agonist) One enantiomer may antagonize the other with no overall effect observed.
Stereoselectivity of one compound can vary for different receptors (+) - 3.23 butaclamol - antipsychotic (-) is almost inactive Eudismic ratio (+/-) is 1250 for D2-dopaminergic, 160 for D1-dopaminergic, and 73 for -adrenergic receptors
Hybrid drugs - different therapeutic activities Antagonist of -adrenergic receptor (-blocker) - triggers vasodilation (-) - 3.25 propranolol (X = NH ) antihypertensive Eudismic ratio (-/+) is 100 But propanolol also is a local anesthetic for which eudismic ratio is 1
Pseudo-hybrid drug - multiple isomeric forms involved in biological activity labetalol - antihypertensive Figure 3.19 R,R- mostly -blocker (eutomer for -adrenergic block) S,R- mostly -blocker (eutomer for -adrenergic block) S,S- and R,S- almost inactive (isomeric ballast)
Racemates as Drugs • 90% of -blockers, antiepileptics, and oral anticoagulants on drug market are racemates • 50% of antihistamines, anticholinergics, and local anesthetics on drug market are racemates • In general, 30% of drugs are sold as racemates Racemic switch - a drug that is already sold as a racemate is patented and sold as a single enantiomer (the eutomer)
Single enantiomer drugs are expected to lower side effects Antiasthma drug albuterol binds to 2-adrenergic receptors, leading to bronchodilation The (R)-(-)-isomer is solely responsible for effects; the (S)-(+)-isomer causes pulse rate increases, tremors, and decreased blood glucose and potassium levels
Sometimes, it is better to use the racemate than one isomer. In the case of the antihypertensive drug nebivolol, the (+)-isomer is a -blocker; the (-)-isomer causes vasodilation by a different mechanism. Therefore, it is sold as a racemate to take advantage of both vasodilating pathways.
Receptor Interaction Figure 3.20 Enantiomers cannot be distinguished with only two binding sites.
Three-point attachment concept Figure 3.21 Receptor needs at least three points of interaction to distinguish enantiomers.
Geometric Isomers The antihistamine activity of (E)-triprolidine (3.35a) is 1000-fold greater than the (Z)-isomer.
Conformational Isomers • Pharmacophore is defined by a particular conformation of a molecule (the bioactive conformation) • The conformer that binds need not be the lowest energy conformer - Binding energy can overcome the barrier to formation of a higher energy conformer
Note that the bioactive conformation bound to the peroxisome proliferator activated receptor gamma (PPAR) is not the lower energy extended conformation. Figure 3.22
If the lead has low potency, it may be because of the low population of the active conformer. If the bioactive conformer is high in energy, the Kd will appear high (poor affinity) because the population of the ideal conformer is low.