Download
1 / 9
90 likes | 261 Vues
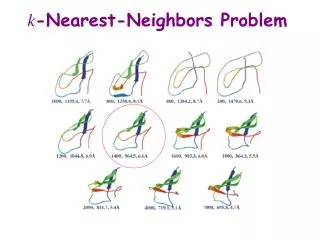
k -Nearest-Neighbors Problem. cRMSD. cRMSD(c,c ’ ) is the minimized RMSD between the two sets of atom centers: min T [(1/n) S i=1, … ,n ||a i (c) – T(a i (c’))|| 2 ] 1/2 where the minimization is over all possible rigid-body transform T. k -Nearest-Neighbors Complexity.

E N D
cRMSD • cRMSD(c,c’) is the minimized RMSD between the two sets of atom centers:minT[(1/n)Si=1,…,n||ai(c)– T(ai(c’))||2]1/2 where the minimization is over all possible rigid-body transform T
k-Nearest-Neighbors Complexity • O(N2(log k + L)) • N number of protein conformations to be compared • K number of nearest neighbors • L time to compare two conformations (cRMSD takes linear time). • Solution reduce L by reducing the number of centers to compare -> m- averaging
m-Averaged Approximation • Cut the backbone into fragments of m Ca atoms • Replace each fragment by the centroid of the Ca atoms
Evaluation: Test Sets[Lotan and Schwarzer, 2003] • FOLDTRAJ random partially unfolded structures -> good correlation with small m (few long segments) • Park-Levitt set [Park et al, 1997] compact native-like structures -> good correlation with large m (many short segments) • Use smaller m on unfolded proteins for greater time savings
Flexible m-averaging • ProteinA 47 residues • 14 < rgyr < 24 • 6 < m < 12 rgyr
Results • Overhead for calculating and m-averaged structures and rgyration too high • Without averaging 28 sec and for all constant m’s 1 min • With flexible average 2 mins 20 sec • Easily fixed by precalculating rgyr and structures
F U Uses
Conclusions • Flexible m-averaging can save time (without sacrificing accuracy?) • Useful for quickly finding k nearest neighbors and building roadmaps • Precalculate m-averaged structures and rgyration for greater speed up