Download
1 / 19
190 likes | 383 Vues
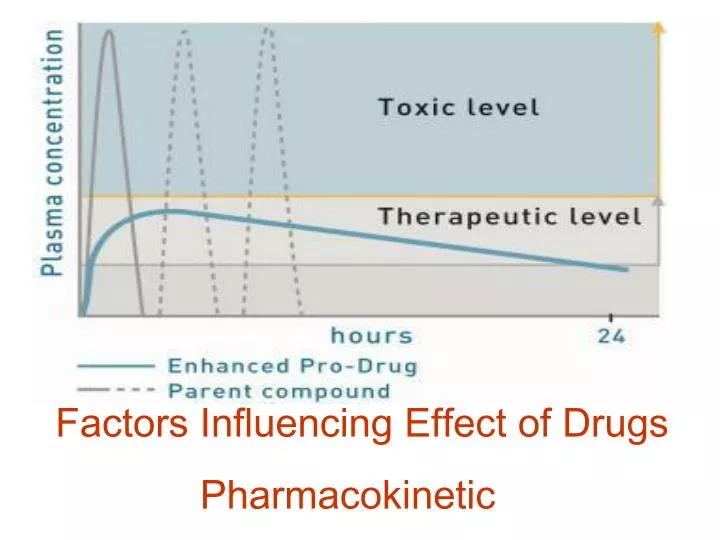
Factors Influencing Effect of Drugs Pharmacokinetic. Factors Influencing Effects of Drugs. external Characteristic of the drug , way of application , doses and their intervals ... They can be easily changed internal Patient´s characteristic Change is impossible or very problematic.

E N D
Factors Influencing Effect of Drugs Pharmacokinetic
FactorsInfluencingEffectsofDrugs • external • Characteristicofthedrug, wayofapplication, doses and theirintervals... • Theycanbeeasilychanged • internal • Patient´scharacteristic • Changeisimpossible or veryproblematic
External Factors Application Per os • Time of drug dissociation, disolution, stability in GIT • pH in GIT, speed of stomach emptying, motility, size of absorption surface, GIT diseases, mesenterial flow • Presence of other substances: drugs (antacids, prokinetics, content of ions – possible chelation), food (possible change of absorption) Intramuscular application - If needed fast effect • If the drug dissolves in GIT (penicilin G) • If high „first pass effect“ (lidocain) • If bad compliance • Influence of liposolubility, pH of solution, muscle perfusion Rectal application • Fast effect, low „first pass“
No irritation of stomach mucous membrane • If patient can´t take drugs per os Pulmonary application • Inhalation of gas, aerosol, particles • Quick effect Application to mucous membrane, skin • Sublingual and buccal (quick absorpcion, lower „first pass“) • Intranasal (peptides – vasopressin, calcitonin) • Transdermal – plasters, gels (nitroglycerin, fentanyl,...) Local application • To mucous membrane of the ear, to conjunctival duffel, to articulation, local application on skin... • Local effect to bronchial stroma ← inhalation
Time of Application Chronopharmacology • glucocortikoids, digoxin, ciklosporin Xenobiotics, drugs Internal Factors Age Children - Absorption (↓ volume of gastric and duodenal liquid, ↓ acidity, ↓ absorption surface) • Biotransformation (immaturity of enzyme systems) • Bigger extracelular space • Incorporation of drug to growing tissues (tetracyclines)
↓ glomerular filtration Geriatric Patients • ↓ acidity, slower motility, longer time to evacuate stomach, venostasis in the splanchnic region - ↓ activity of biotransformating enzymes, ↓ perfusion of liver • ↓ concentration of binding proteins • ↓ weight of the body, slower circulation • ↓ of kidney function Gender Women • ↓ absorption surface • Competition of estrogens and progesteron with metabolism • ↓ glomerular filtration, ↓ weight, ↑ ratio of body lipids • Choice of drugs with respect to gravidity and lactation
Patologic state absorption • GIT diseases (quicker or slower pasage, surgery, inflammation in the area of application) distribution • States connected with hypoalbuminemia (nephrotic syndrome, deffect of proteosynthesis in liver) biotransformation • Diseases of liver parenchyma (changes of metabolisation, presystemic elimination) excretion - Kidney deffects, changes of inner space
Genetic factors genetically dependent changes: • pharmacodynamics - resistance to cumarines, decreased effect of β-blockers among blacks • pharmacokinetics - biotransformational polymorfism (slow, fast acetylators, hydrfoxylators)
Pharmacokinetics Study of drug concentration development and its metabolites in time • Working out of suitable matematic models for interpretation of these datas Drug in organism • Absorption • Distribution • Elimination (biotransformation + excretion)
Transport through membranes • Always depends on time • Speed of molecule transport through membranes + speed of molecule changes (biotransformation) → concentration in given place • Pasive transport – in the way of gradient (concentration/electric) • Free transfer • Transfer through ion channels • Basic diffusion through lipid membrane • Transfer through intercellular pores • Facilitated diffusion (through carrier, without energy need) • Active transport – against the way of gradient, carrier and energy needed • Endocytosis – carries out vakuolar aparatus of some cells (e.g. enterocytes)
Possibility of molecular transfer through membranes by passive transport depends mainly on the size, ionisation and hydrophility/lipophility of the molecule • Transfer is easier and faster for smaller, electrically neutral and more lipophilic molecules Absorption - Depends on application form • At i.v. application practically no absorption, drug is instantly distributed in systemic circulation → the fastes possible effect • The most common is application per os • Factors influencing GIT absorption: pH (in stomach better absorption of acids, in small intestine of weak basis), perfusion, motility of intestine, patologic changes of mucosal membrane (malabsorption), presence of ensymes, other substances and drugs Distribution • To different compartments • Binding to plasmatic proteins → fixed/not bounded fraction of drug, effective can be only not bounded fraction!
Factors influencing distribution: application form, binding to plasmatic proteins, hydrophility/lipophility of surrounding, first pass effect, barriers (hematoencephalic, placenta), special transport mechanisms (transferin) • Bioavailability – part of substances that reaches systemic circulation • I.v. application → bioavailability=1 (100 %) Biotransformation • Many parts of organism, main organ liver, also kidneys, intestine, lungs • Goal: such a change of applied substance, so it would be eliminated as fast as possible • Can arise: non-active metabolite; metabolite with stronger effect (change from „prodrug“ to effective substance); toxic metabolite with qualitatively changed effect
Basically 2 phases of changes – participating are many ensymes: I. phase – oxidation and reduction, hydrolysis → ↑ sollubility in water oxidation– system of composite oxidases, mikrosomal ensyme system of cytochrome (CY) P450 – many isoensymes hydrolysis – non-specific esterases II. phase – conjugation reactions - sulphatation and glukuronidation - great influence has induction/inhibition of ensymes of system CYP 450 Excretion - Many ways, main organ kidneys - The most important processes influencing renal excretion: glomerular filtration passive backward tubullar difusion active backward tubullar resorption active tubullar secretion - pH of urine (with more acid urine are better excreted basic molecules, with more basic acid molecules)
quantitative description of pharmacokinetic actions uses ph. parameters primary pharmacokinetic parameters secondary Primary ph.p. - directly determined by physiological variables distributory volume – counted from concentration of substance in blood and expresses volume that would occupate particles of the whole given dose of substance, if they would be „inflated“ or „diluted“ the same as in tested sample Vd = D/C0 - only fictive value – it gives idea about the distribution of substance in organism rate constant of absorption (ka) a elimination (ke) inform about speed of these procceses clearance – volume of blood (resp. fraction of Vd), which per unit time fictively completely clears from applied substance
clearance can be counted for the whole organism or for individual organs and compartments • hepatic clearance informs about biotransformation ratio to elimination of drug • renal clearance determines how to reduce doses at kidney disorders Secondary ph. p. - derived from primary ph. parameters biologic half-life – time, after which concentration (ammount) of given drug is reduced to 50% - plasmatic half-life – time, after which is reduced to 50% level of drug in blood - elimination half-life – time, after which is eliminated from organism 50% of applied drug - equations: t1/2 = ln2/ke and t1/2 = ln2.Vd/CL, or approximately t1/2 = 0,7/ke and t1/2 = 0,7.Vd/CL - biologic half-life values are foundation for determining of intervals between doses!! area under the curve (AUC) concentration of drug gives an idea about changes
of concentration in time bioavailability of drug form – determines quality of drug form plasma level – informs about concentration of drug in circulation → at many types of substances determines effect • Single dose – bolus • Most drugs are given as repeated doses • Too low doses – needed therapeutic effect isn´t reached, too high doses – risk of cumulation • Administering of the same therapeutic doses – level of drug progressively increases, till amout of absorbed and eliminated substance is equal → steady state • This occurs after approximately 4 elimination half-lifes!