
PROGRAMS
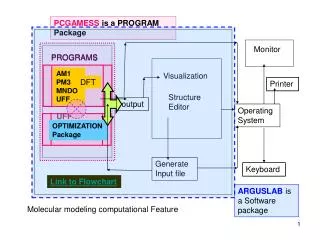
PCGAMESS is a PROGRAM Package. PROGRAMS. AM1 PM3 MNDO UFF. AM1 PM3 MNDO UFF. DFT. DFT. ARGUSLAB is a Software package. Monitor. PROGRAMS. Visualization. Printer. Structure Editor. output. Operating System. OPTIMIZATION Package. Generate Input file. Keyboard.

PROGRAMS
E N D
Presentation Transcript
PCGAMESS is a PROGRAM Package PROGRAMS AM1 PM3 MNDO UFF AM1 PM3 MNDO UFF DFT DFT ARGUSLAB is a Software package Monitor PROGRAMS Visualization Printer Structure Editor output Operating System OPTIMIZATION Package Generate Input file Keyboard Link to Flowchart Molecular modeling computational Feature
Molecular Modelling Essentially has a Quantum mechanical formalism to calculate the energy of a system with atomic coordinates as inputs. This is referred to as the Single point Energy Calculation. The Geometry Optimizing Capability uses this single point calculation program with an Optimization algorithm: Geometry Optimization can be effected by starting with an initial guess structure; and Vary the atomic coordinates in the molecule by a (an infinitely) small amount and find out whether the energy of the molecule increases or decreases. If the energy decreases, continue to Vary the coordinates in the same sense by finite amount and calculate the single point energy. If the energy starts increasing at any stage, then vary the coordinates in the opposite direction. The inference on small variation can be processed to apply a larger change and calculate the Energy with the resulting set of coordinates. The progress of optimization can be continuously monitored by sending the output at every stage/step to the monitor as the calculation proceeds.
Thus the Single point Energy calculation must be by a relevant and well suited algorithm. This is the FORMALISM part which gives rise to the variations in the Calculation Methods (AM1, UFF, DFT etc.,) and different levels of approximations would be convenient depending on the system and properties calculated. Energy is one of the property invariably required for calculating any other property. When the calculations proceed by calculating all the relevant integrals using the mathematical functions describing the electron orbitals, then it is referred to as Abinitio method. If some of the integrals are identifiable with experimentally measureable properties and these values are used in place of evaluated integrals, then it becomes a semi empirical method. Such Parametrization of several types and to different extents are in vogue.
START YES NO PRINT INPUT FILE READ IN YES (iteration No. ‘n’ in general) Is the RMSD at this stage Less than the desired limit of Convergence Criterion? (Increment iteration number due) Option for which is the Quantum Mechanical Method to Use.. Single point Energy, or Geometry Optimiztion etc., Initial set of atomic coordinates Single point energy by the Quantum Chemical method Opted Read Initial set of Atomic Coordinates list Line Search method OPTION iteration No: ‘0’ Geometrical Optimization? Iteration No. ‘0’ Calculate Single point energy with modified Coordinates NO Calculate the necessary Gradients and RMSD: Apply Correction to the existing coordinate set Iteration ‘n+1’ STOP END Click to start AM1 PM3 MNDO DFT UFF Click Click Click Click Optimization Routine CLICK HERE For elaboration Click CLICK HERE to get back to slide#1 Click Click Click Click Click Click Click Visualization Monitor Click Click Click Click Click Click Click Click

















