Proteins ? 1D 3D
400 likes | 602 Vues
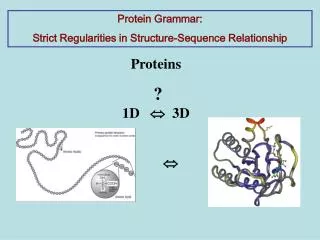
Protein Grammar: Strict Regularities in Structure-Sequence Relationship. Proteins ? 1D 3D. . The main rule of protein Sequence – Structure relationship :.

Proteins ? 1D 3D
E N D
Presentation Transcript
Protein Grammar: Strict Regularities in Structure-Sequence Relationship Proteins ? 1D 3D
The main rule of protein Sequence – Structure relationship : The amino acid sequence alone is sufficient to determine a protein's structure. Christian Anfinsen, 1961 The Nobel Prize in Chemistry 1972
From The Anfinsen rule follows: 1) Protein folding is a physical problem but not biological (?) Blue Gene is an IBM Research project dedicated to exploring thefrontiers in supercomputing: in computer architecture, in the software required to program and control massively parallel systems, and in the use of computation to advance our understanding of important biological processes such as protein folding The Blue Gene/L machine has a peak speed of 596 Teraflops ab initioapproach
From The Anfinsen rule follows: Thus a structure can be determined by analogy with known protein structures of similar sequences. The idea that sequence similarity translates into structural similarity underlies most modern high-accuracy algorithms of structure prediction 2) it is to be expected that similar sequences would encode similar structures homology approach
similar ? similar ? Amino acid Sequences 3D Protein structures ? relationship ? We need to define similar structures ? similar sequences ?
Similarity of structures ? The first main rule of Protein structures Fundamental Units of Protein Structure Hydrogen bonds form helices - alpha form and beta-form W. Astbury (1930s), L. Pauling (1939-1951 ) hydrogen atoms oxygen A. alpha-helix B. beta-sheets
Beta Sandwich-like Proteins two main -sheets packed against each other.
beta Sandwich-like Proteins 1 2 3 8 9 N…——>…——>…——>……——>…——>… The goal is to find Are any rules in the packing of strands in Sandwich-like structure? Folding pattern
Part I. Folding pattern of SSS Stage 1: Collection All Sandwich Proteins are collected from SCOP and CATH databases. We analyzed 81 superfamilies and 177 families ~ 8,000 structures.
Stage 2: Structure description Description of structures in strands: .5-4-8-9-2 . 6-7-3-1 For over 40 years, researchers have looked at how Secondary structural elements– strands and helices assemble into structure Secondary structure motif Stage 2: Classification Description of SSS in strands: 5-4-8-9-2 6-7-3-1 5-4 8-9 2 6-7 3 1 We introduced a new supersecondary structure unit –”strandon” - a set of the maximum number of consecutive strands, which are connected in the sequential order by hydrogen bonds in 3D structure. Description of SSS in strandons: IV VI II V III I
1 2 3 8 9 N…——>…——>…——>……——>…——>… We introduced a new supersecondary structure unit –”strandon” - a set of the maximum number of consecutive strands, which are connected in the sequential order by hydrogen bonds in 3D structure. Strandon i i +1 Supersecondary structure elements
Description of structures in strandons: IV VI II V III I Now we suggest to investigate how Supersecondary structure elements - strandons assemble into structure.
Stage 3: SSS classification All proteins were described in STRANDON’ notation I III (9 1 2 3) (6) I III IVII MOTIF (8 7)(4 5) IV II supermotif
This is the basis of novel hierarchical classification in the Supersecondary structures (SSS) database supermotif motif protein structure
I III V I III II VI IV IV II I III I III VII V II IV II VIII IV VI I V III III I V VI II IV II IV VI 6 SUPERMOTIFS describe ~ 90% of all sandwich structures.
Stage 4. SSS regularities: Analysis of all supermotifs in the SSS database led us to the discovery of the rule of Supermotifs
The Rule of Supermotifs – Rules of arrangement of strandons in the twomain beta sheets. 95% of all structures obey the rule of supermotifs. K=1, N=4K=4, N=4K=3, N=6 I IIIIV IIIII V I II IVI IIIIV II VI
The rule of supermotifs dramatically restricts the number of permissible arrangements of strandons. Analysis of observed arrangements of strands within the strandons leads us to formulate the rule of motifs.
Rule of Motifs For two neighboring strandons in a sheet, or at the edges of the same side of two beta-sheets, the strands’ numbers in these two strandons will increase in opposite directions.
The Rule of Motifs • ( ordering ofstrands within the strandons ) • held true for all strandons in 82% of analyzed protein domains. • In 12 % of the structures the ordering of strands is obeys the Rule of Motifs in all strandons but ONE strandon. • (82 + 12) % domains
Question: How strands come together in structures of Sandwich proteins ? Answer: Structures of beta-sandwich proteins are governed by well-defined rules: the Rules of Motifs, and the Rules of Supermotifs These rules describe the Folding Patterns End Part I - Folding patterns
Protein structures similarity definition: Proteins with the same secondary structure motif and the same orientation of strands in two beta sheets have similar protein structures. 1 2 4 1 2 4 Sheet A Sheet A Sheet B Sheet B 8 7 5 3 8 7 5 3
Part II - Sequence patterns The main problem: How to extract a structure information from the sequence, and how to reconstruct tertiary structure? similar Amino acid Sequences 3D Protein structures ? relationship ? • Idea: • Collect all proteins with similar structures. • Find proteins with non-similar sequences (from different protein families. • Extract common sequence regularities if exist
Part II - Sequence patterns The main problem: How to extract a structure information from the sequence, and how to reconstruct tertiary structure? non similar similar Amino acid Sequences 3D Protein structures ? relationship ? • Idea: • Collect all proteins with similar structures. • Find proteins with non-similar sequences (from different protein families. • Extract common sequence regularities if exist
SSS database motif: sheetI: 1 2 5 4 sheetII: 7 6 3 This motif describes proteins from 3 families. Sequences from different families are strongly dissimilar. Sequence alignment reveals 1-4% of identical residues. Alignment 2 sequences: EMBOSS NeedleprogramBlast program #1: 1f42 #2: 1oke Identity: 1.9% No significant similarity found
1) Collect all proteins with identical SSS 2) Find proteins with non-similar sequences (from different protein families. ? 3) Extract common sequence regularities if exist Hypothesis Proteins with similar structures share a unique set of residues - ‘Structure -determining residues’ - even though they may belong to different protein families and have very low sequence similarities
The problem: the widely used alignment algorithms - PSI-BLAST, NEEDLE - are not applicable to sequences with very low sequence similarity. Therefore for comparison of sequences of proteins that share same SSS, we developed a new algorithm of structure-based multi-sequence alignment. Alignment 2 sequences: EMBOSS NeedleprogramBlast program #1: 1f42 #2: 1oke Identity: 1.9% No significant similarity found
New SSS-based multi-sequence alignment algorithm The main feature of this algorithm: Units of alignment are individual strands and loops, rather than whole sequences. Strands Alignment of strands in a beta sheet is based on hydrogen bond contacts. No gaps are allowed within strands. Loops Local alignment of each loop separately With gaps.
Select the best variant with max numbers of conserved positions (?)
conserved positions (?) 20 amino acids are divided into 2 groups: Q, E, R,T, Y, P, S, D, G, H, K, N – HYDROPHILIC residues W, I, A, F, L, C, V, M - HYYDROPHOBIC residues matching position is conserved if all (almost) residues in this position belong to one of these groups in all proteins.
Proteins sheetI: 1 2 5 4 sheetII: 7 6 3 3 protein families 71,786 sequences 601 sequences
10 representative sequences . for alignment 1) ••• 2) ••• 3) ••• 4) ••• ••• 10) •••
Set of Structure-determining residues 30 conserved positions: 19 - hydrophilic and 11 - hydrophobic Question: Are residues at 30 conserved positions specific and sensitive?
Testing specificity and sensitivity. Are the residues at conserved positions the SSS-determining residues? How many proteins describes the set of residues at conserved positions in 3 protein families (true positives) and in other proteins (false positives)? Answer : EMBOSS/Preg program revealed 304 - true positives (of 601 proteins) Not good! 0 - false positives Good! 71,786 sequences
Refining the definition of SSS-determining residues 1) Find additional residues at the conserved positions: gradually add residues respectively to the hydrophobic and hydrophilic conserved positions and test how a new “extra” residue affects on specificity and sensitivity. 2) Vary the distance between a conserved position in a strand and a conserved position in a loop, and between conserved positions within loops. Result: EMBOSS/Preg program with the new set of residues revealed: 573 - true positives (of 601 proteins) 0 - false positives
The set of SSS-determining residues with a single mismatch position. To identify additional true positives any residue is allowed at any single position. Result: EMBOSS/Preg program with the new set of residues revealed: additional 18 true positive sequences, and no false positives. Our analysis found that the remaining 6 sequences have 2 mismatching positions. Important conclusion: substitution of a hydrophilic for a hydrophobic residue, or vice verse is allowed at just 1-2 conserved positions.
Conclusion sequence structure . relationship 1) Sequence of amino acids defines 3-D structure; 1-D 3-D 2) Similar 3-D structures have an unique set of structural determinants. 3-D 1-D Protein sequence-structure relationship is reciprocal.
Acknowledgements Professor Israel Gelfand, Dr. Yih-Cheng Chiang Dr. Cyrus Chothia SSS database