Download

1 / 18
180 likes | 301 Vues
crowmether-host1.ppt. Chiral Recognition by NMR Spectroscopy- A Theoretical approach. Illustrating modeling strategies with organic molecules with overtones for Modeling of Bio-molecules An Abstract S.Aravamudhan This material may be available as Internet Resource:

E N D
crowmether-host1.ppt Chiral Recognition by NMR Spectroscopy- A Theoretical approach. Illustrating modeling strategies with organic molecules with overtones for Modeling of Bio-molecules An Abstract S.Aravamudhan This material may be available as Internet Resource: Display the web subdirectory: http://www.ugc-inno-nehu.com/CRNMR/ • Some of the linked files in this .ppt file require the Software “ARGUSLAB” be installed in your system. Download the MS Windows installer “setup.exe” file by clicking on the link below: • http://www.ugc-inno-nehu.com/arguslab/ And download the all the contents of this directory into the same and single folder in the resident disc of the P.C. for the hyperlinks in the presentation file to display the appropriate file. Aravamudhan: Chiral Recognition by NMR
The Chiral recognition by NMR require: NMR spectrum of the Host molecule Slide#13 to 15 NMR spectrum of the Guest (Chiral) molecule Slide#18 NMR spectrum of the Host-guest complex. Identifying the spectral lines/region which indicates the dependence on the complexing by the guest. Slide#3 And, monitoring the indicator peaks with the concentration of the Guest molecule. This means a theoretical calculation would require calculation of the NMR of the complex molecule for various Stoichio metric proportions and hence Host to Guest molar ratios - 1:2, -1:1, - 2:1, -etc ., to be specifying a few typical ratios. To cover non-integral ratio values, the required integral number of cluster molecules would demand handling large sized molecular cluster than for the above simple integral ratios. For such clusters Host-host, guest-guest interactions and changes in structures of host and guest molecule structures would be factors to reckon with. This would mean the super molecular system to handle would increase in size depending on the complex as compared to the size of the guest molecule alone or the host molecule alone. The increase in computation time, and while trying to optimize the time factor, what options could be exercised on the theoretical method and basis sets, must be spelt out with the corresponding advantages/disadvantages. S.Aravamudhan CRNMR
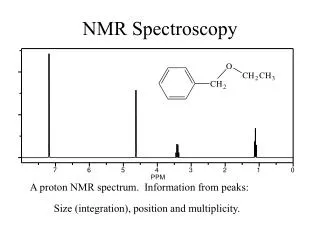
Increasing amount of Guest molecule Return to Slide#2 Variation of NMR spetrum with Concentration of guest The Full publication by Koylu et al The Full publication by Koylu et al The Full publication by Koylu et al S.Aravamudhan CRNMR
The enumeration of Host Molecules considered and the computationally convenient system: A model Host to begin with. S.Aravamudhan CRNMR
These 42 atoms (or more ) ether-molecule have reasonably simple NMR structures. This implies the high symmetry of the molecule ; and a small guest molecule, even if it has chiral centre, does not display much distinctive features of interaction with the host for the guest R and S conformations. Thus, it becomes necessary to find appropriate derivatives of the crown ether for the CR properties of the complex, resulting in much larger sizes for the molecules and the molecule-clusters which is an important criterion for the theoretical calculations (computation). This invariably is a criterion while optimizing (larger biological) macro-molecules and hence mostly for time factors the MM is used. MM is good enough for handling inter-residue interactions, and intermolecular interactions for clusters. However, intra-molecular changes during molecular cluster optimization are seemingly necessary to reckon with, hence a hybrid method MM/QM could be preferable. An assessment of this type and how to go about discerning the various requirements can be illustrated, and becomes better appreciable with instances at smaller molecule levels D3d S.Aravamudhan CRNMR
From these results it is evident that the popularly used Crown Ether system is too large for computational investigations Webmo Jobs links: 107509 107510 107511 107512 Deleting the Guest molecule from the complex & Calculating NMR S.Aravamudhan CRNMR
The charge densities at O11 and the nearby protons show considerable variation due to the QMM used. The charge/shift trends on proton (H12 and) H13 seem to indicate a possibilities of through space contributions and not only through bond for the chemical shifts. Much careful monitoring of such trends would be required is an obvious consequence. GO with Gaussian by Abiitio HF/STO3G Gaussian Follwoed by chemical shifts calcualtion Job 104673 (5.3Secs) down field up field This slide material highlights the differences that can arise due to variation of the QM method: level of approximations used GO with ARGUSLAB by semiempirical QMM: PM3 Follwoed by chemical shifts calcualtion Job 104672 (5.3secs) The image file: 18-Mod29-GObyHFsto3g-GObyPM3-NMRbyHFsto3g-65pc.JPG S.Aravamudhan CRNMR
Return to Slide#2 S.Aravamudhan CRNMR
The enumeration of Guest Molecules considered and the computationally convenient system: A model Host to begin with. S.Aravamudhan CRNMR
Host deleted in Str. Editor after docking, and guest NMR calculated: Thus indicating a change in the guest molecule electronic structure during the Docking The ligand molecules (R and S-guest) –GO and NMR calculated: Host-guest complex-GO : subsequent simple deletion of the host: and the Guest molecule NMR calculated Return to Slide#2 S.Aravamudhan CRNMR