Download
1 / 18
180 likes | 458 Vues
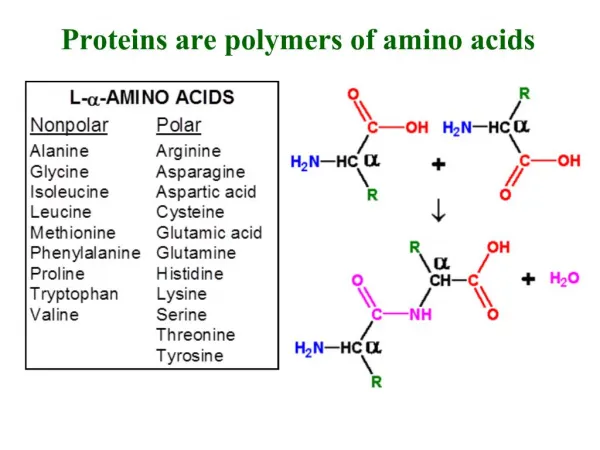
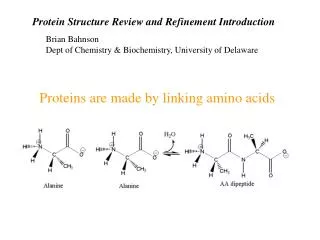
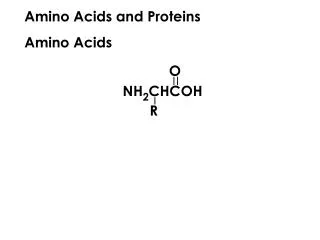
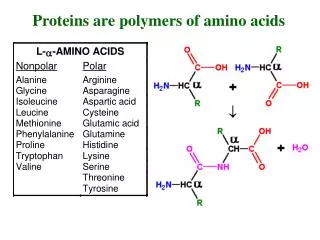
R. N. Proteins are built of L-amino acids. 20 different amino acids are encoded by specific DNA base triplets. The amino acids are linked together by amide bonds. Proteins are linear chains of amino acids. Peptides are short proteins (< 50 residues)

E N D
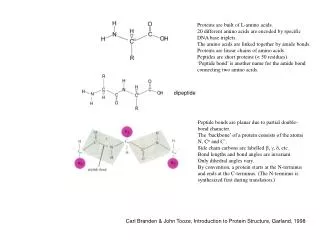
R N Proteins are built of L-amino acids. 20 different amino acids are encoded by specific DNA base triplets. The amino acids are linked together by amide bonds. Proteins are linear chains of amino acids. Peptides are short proteins (< 50 residues) ‘Peptide bond’ is another name for the amide bond connecting two amino acids. dipeptide Peptide bonds are planar due to partial double- bond character. The ‘backbone’ of a protein consists of the atoms N, Ca and C’. Side chain carbons are labelled b, g, d, etc. Bond lengths and bond angles are invariant. Only dihedral angles vary. By convention, a protein starts at the N-terminus and ends at the C-terminus. (The N-terminus is synthesized first during translation.) Carl Branden & John Tooze, Introduction to Protein Structure, Garland, 1998
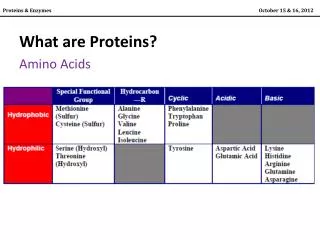
glycine, Gly, G alanine, Ala, A hydrophobic: A I L M F P W V positively charged: R K (H) negatively charged: D E polar: C N Q S T Y tiny: G arginine, Arg, R aspartic acid, Asp, D asparagine, Asn, N cysteine, Cys, C glutamine, Gln, Q glutamic acid, Glu, E isoleucine, Ile, I leucine, Leu, L histidine, His, H lysine, Lys, K methionine, Met, M phenylalanine, Phe, F proline, Pro, P serine, Ser, S threonine, Thr, T tryptophan, Trp, W tyrosine, Tyr, Y valine, Val, V
Hydrophobic cores are tightly packed The interior of protein structures is tightly packed. Water is excluded, except for very few buried hydration water molecules. Almost all residues in the interior are hydrophobic or, at least, uncharged. Charged residues are almost always on the protein surface. The same rules apply to protein-protein binding surfaces. Regular secondary structures form, because amide groups are polar, seeking H-bonding partners when buried in a hydrophobic environment. CPK model of ubiquitin yellow: hydrophobic grey: polar but uncharged blue: positively charged red: negatively charged green: backbone atoms Backbone H-bonds Only backbone and hydrophobic sidechains retained
Since peptide bonds are planar (and virtually always ‘trans’), the backbone conformation of each amino acid is determined by only two dihedral angles, f and y. Knowledge of the f/y pairs of each residue is sufficient to define the 3D structure of the entire backbone!
Ramachandran plot a-helix b-sheet left-handed a-helix non-Gly residues Gly The Ramachandran plot displays experimentally observed f/y combinations (one dot per residue). Steric clashes between the side chains of neighboring amino acids limit the accessible conformational space. Glycine can access larger regions in the Ramachandran plot than residues with longer side chains.
Primary, secondary, tertiary, quarternary structure Primary structure = amino acid sequence Secondary structure = helices, sheets, turns (i.e. regular sub-structures defined by H-bonds between backbone amides) Tertiary structure = 3D structure Quarternary structure = complex between different protein molecules (e.g. dimer, trimer, tetramer) 2 Cys residues can form a disulfide bridge: -CH2-SH + 1/2 O2 -CH2-S-S-CH2- + H2O
a-helix: 3.6 residues per turn, H-bonds between residues i and i+4
antiparallel b-sheet parallel b-sheet in both types of b-sheets, the side-chains point alternatingly above and below the plane of the sheet
mixed b-sheet example: thioredoxin
Leventhal’s paradox Assume a small protein with 100 amino acids, each one of them can access 3 different conformations 3100 = 5 x 1047 conformations Fastest motions 10-15 sec, so sampling all conformations would take 5 x 1032 sec 60 x 60 x 24 x 365 = 31536000 = 3.1536 x 107 seconds in a year Sampling all conformations will take 1.6 x 1025 years, much longer than the age of the universe In nature, proteins fold correctly within seconds! The 3D structure is unambiguously encoded in the amino-acid sequence, but protein structures are very hard to predict from amino acid sequence, unless the structure of a similar protein (> 20% amino-acid sequence identity) is known. Different proteins fold by different, unpredictable mechanisms (some of them even need helper proteins (“chaperones”) to fold. The current picture is that of a folding funnel, where the vertical axis displays energy and the width of the funnel represents the accessible conformational space.
Secondary structure prediction PHD is best: http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_phd.html Expected accuracy: 72% 10 20 30 40 50 60 70 | | | | | | | MGRARDAILDALNLTAEEKLKKPKLELLSVPLREGYGRIPRGALLSMDALDLTDKLVSFYLETYGAELTA CCHHHHHHHHHHHHHHHHHHHHHHHHHhhcchhHhcCcCcHHHHHhcCHHHHHHHHHHHHHHHHhHHHHH NVLRDMGLQEMAGQLQAATHQ HHHHHHHHHHHHHHHHHHccC Sequence length : 91 PHD : Alpha helix (Hh) : 77 is 84.62% 310 helix (Gg) : 0 is 0.00% Pi helix (Ii) : 0 is 0.00% Beta bridge (Bb) : 0 is 0.00% Extended strand (Ee) : 0 is 0.00% Beta turn (Tt) : 0 is 0.00% Bend region (Ss) : 0 is 0.00% Random coil (Cc) : 14 is 15.38% Ambiguous states(?) : 0 is 0.00% Other states : 0 is 0.00% MGRARDAILDALNLTAEEKLKKPKLELLSVPLREGYGRIPRGALLSMDALDLTDKLVSFYLETYGAELTANVLRDMGLQEMAGQLQAATHQ CCHHHHHHHHHHHHHHHHHHHHHHHHHhhcchhHhcCcCcHHHHHhcCHHHHHHHHHHHHHHHHhHHHHHHHHHHHHHHHHHHHHHHHccC --HHHHHHHHHHH—HHHHHHHHHHH---------------HHHHHH—HHHHHHHHHHHH—HHHHHHHHHHHHHHHH---HHHHHHHHHH--
ca. 20% sequence identity can still result in similar 3D structure (statistical sequence identity: 6%) Bars identify side-chains with less than 5% solvent accessibility in PYRIN domain Boxes delineate helix boundaries Vertical grey-shading: conserved core residues PYRIN DED CARD DD Death domains (DD), death effector domains (DED), caspase activation and recruitment domains (CARD) and PYRIN domains form 4 branches of the death domain superfamily, i.e. their 3D structures are related while their sequence similarity is limited (J. Mol. Biol. 332, 1155 (2003)).
3D structure prediction Swiss-Model: www.expasy.org/swissmod/SWISS-MODEL.html If a template structure with >25% amino acid sequence identity is available in the Swiss-Model database, only the amino acid sequence needs to be submitted. Otherwise, the 3D coordinates of the desired template structure must be submitted too. Length of target sequence: 91 residuesSearching sequences of known 3D structuresNo suitable target found ==> Exit Swiss-Model and any other modelling software (best regarded is Modeller, www.salilab.org/modeller/modeller.html) depend crucially on the sequence alignment. Any model has the same coordinates for backbone and Cb atoms as the template. Insertions and deletions are handled ‘gentlemanly’.
Identification of similar structures with Dali FSSP FAMILIES OF STRUCTURALLY SIMILAR PROTEINS, VERSION 1.0 (Apr 1 1995) CREATED Fri Nov 1 01:15:14 GMT 2002 for dali on sputnik2-node68.ebi.ac.uk METHOD Dali ver. 2.0: Holm, L., Sander, C. (1993) J.Mol.Biol. 233,123-138 DATABASE 3241 protein chains PDBID 6340 HEADER Structure from MOLMOL COMPND pyrin SOURCE AUTHOR SEQLENGTH 90 NALIGN 54 WARNING pairs with Z<2.0 are structurally dissimilar ## SUMMARY: PDB/chain identifiers and structural alignment statistics NR. STRID1 STRID2 Z RMSD LALI LSEQ2 %IDE REVERS PERMUT NFRAG TOPO PROTEIN 1: 6340 6340 23.2 0.0 90 90 100 0 0 1 S pyrin 2: 6340 1a1z 8.5 2.2 79 83 19 0 0 5 S fadd protein fragment (fas-associating death domain-con 3: 6340 1ich-A 7.3 2.3 75 87 19 0 0 6 S tumor necrosis factor receptor-1 fragment (tnf-1) Muta 4: 6340 3ygs-P 6.9 2.5 78 97 19 0 0 6 S apoptotic protease activating factor 1 fragment procasp 5: 6340 1ngr 6.6 2.6 73 85 10 0 0 5 S p75 low affinity neurotrophin receptor fragment 6: 6340 1dgn-A 5.9 2.8 77 89 13 0 0 6 S iceberg (protease inhibitor) fragment 7: 6340 1cy5-A 5.5 3.4 79 92 15 0 0 6 S apoptotic protease activating factor 1 fragment (apaf-1 8: 6340 1d2z-B 5.0 2.8 80 150 9 0 0 7 S death domain of pelle death domain of tube 9: 6340 3crd 4.6 3.0 75 100 15 0 0 7 S raidd fragment 10: 6340 1ddf 4.3 2.9 73 127 12 0 0 4 S fas 11: 6340 1g71-A 4.0 3.1 68 344 12 0 0 7 S DNA primase 12: 6340 1au7-A 3.8 3.5 62 130 6 0 0 7 S pit-1 fragment (ghf-1) Mutant biological_unit DNA 13: 6340 1d2z-A 3.7 2.5 63 102 5 0 0 6 S death domain of pelle death domain of tube 14: 6340 1dly-A 3.5 3.2 65 121 12 0 0 5 S hemoglobin http://www.ebi.ac.uk/dali/ alternative: http://cl.sdsc.edu/
Large ribosomal subunit from Haloarcula marismortui Science 289, 905 (2000)
From Structure to Function Convergent evolution: the overall structures of chymotrypsin and subtilisin are very different, but the catalytic triade (Asp, His, Ser: side-chains shown in blue) is conserved chymotrypsin subtilisin
A mechanism for the evolution of proteolytic function FMN-binding protein from Desulfovibrio vulgaris protease from hepatitis virus Views differ by a 90o rotation around a vertical axis. Catalytically active residues shown as spheres. The structure is composed of two domains, each of which is similar to the FMN-binding protein. The binding site of FMN is at the site corresponding to the substrate binding site in the protease. Two orthogonal views of chymotrypsin backbone allosteric regulation site* Chymotrypsin consists of two subdomains of similar structure. The active site is at the interface. The residues of the catalytic triade are contributed by both domains. substrate binding site • Chymotrypsin is activated by proteolytic cleavage of the N-terminal end, resulting in altered binding of the new N-terminus to the • C-terminal domain. Nat. Struct. Biol. 4, 975 (1997)
Steps for evolution of proteolytic function: • a primordial peptide-binding protein (similar to the FMN-binding protein) • gene duplication, resulting in 2 domains linked by a polypeptide chain • any proteolysis, even if inefficient, decreases cooperativity of binding, • hence peptide fragments dissociate, enabling capture of new, uncleaved peptide gene duplication Proteolysis could occur, if the correct residues approach the peptide in the cleft between both domains. No increased chance for proteolytic activity If this evolutionary pathway is correct, what happened to the peptide binding site that is not at the interface between the two domains? It became an allosteric regulation site, i.e.is still a peptide binding site in a way.