Advances in Sequence Analysis: Parsimony, Similarity, and MCMC Approaches
This paper discusses various methods for sequence analysis, including parsimony, similarity, and optimization techniques such as MCMC. We explore the TKF91 substitution/indel process, demonstrating its application to models like GT-CAT, GTTGGT, GT-CA, and CT-CA for improved sequence alignment. Emphasis is placed on practical implementations and phase analyses, along with statistical performance evaluations. The results contribute to further understanding of evolutionary relationships and provide insights into the efficiency of sequence comparison algorithms over time.

Advances in Sequence Analysis: Parsimony, Similarity, and MCMC Approaches
E N D
Presentation Transcript
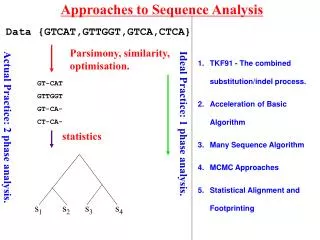
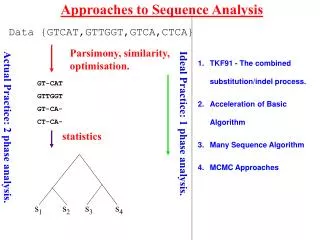
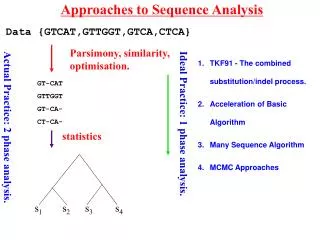
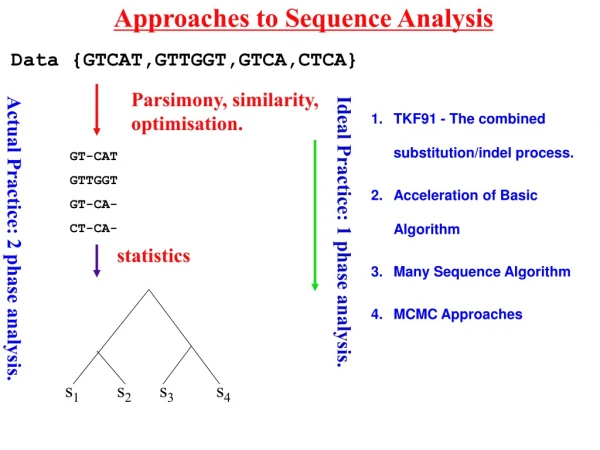
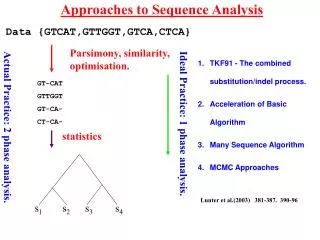
Approaches to Sequence Analysis Data {GTCAT,GTTGGT,GTCA,CTCA} Parsimony, similarity, optimisation. TKF91 - The combined substitution/indel process. Acceleration of Basic Algorithm Many Sequence Algorithm MCMC Approaches GT-CAT GTTGGT GT-CA- CT-CA- Ideal Practice: 1 phase analysis. Actual Practice: 2 phase analysis. statistics Lunter et al.(2003) 381-387. 390-96 s1 s2 s3 s4
T= 0 # - - - ## # # # T = t # # # # s1 r s2 s1 s2 s1 s2 Thorne-Kishino-Felsenstein (1991) Process A # C G * • (birth rate) < m(death rate) 1. P(s) = (1-l/m)(l/m)l pA#A* .. *pT #T l =length(s) 2. Time reversible:
# - - - - - # # # # k * - - - - * # # # # k l & m into Alignment Blocks A. Amino Acids Ignored: # - - - ## # # k e-mt[1-lb](lb)k-1 [1-lb-mb](lb)k [1-lb](lb)k p’k(t) pk(t) p’’k(t) b=[1-e(l-m)t]/[m-le(l-m)t] p’0(t)= mb(t) B. Amino Acids Considered: T - - - RQ S W Pt(T-->R)*pQ*..*pW*p4(t) 4 • T - - - - • R Q S WpR *pQ*..*pW*p’4(t) • 4
Dpk = Dt*[l*(k-1) pk-1 + m*k*pk+1 - (l+m)*k*pk] Dp’k=Dt*[l*(k-1) p’k-1+m*(k+1)*p’k+1-(l+m)*k*p’k+m*pk+1] Dp’’k=Dt*[l*k*p’’k-1+m*(k+1)*p’’k+1- [(k+1)l+km]*p’’k] Differential Equations for p-functions # - - ... - # # # ... # # - - - ... - - # # # ... # * - - - ... - * # # # ... # Initial Conditions: pk(0)= pk’’(0)= p’k (0)= 0 k>1 p1(0)= p0’’(0)= 1. p’0 (0)= 0
Basic Pairwise Recursion (O(length3)) i j Survives: Dies: i-1 i i-1 i j-1 j j i-1 i i j-2 j i-1 j j-1 …………………… …………………… …………………… e-mt[1-lb](lb)k-1, where …………………… …………………… b=[1-e(l-m)t]/[m-le(l-m)t] 0… j (j+1) cases 1… j (j) cases
Basic Pairwise Recursion (O(length3)) survive death j (i-1,j) j-1 (i-1,j-1) Initial condition: p’’=s2[1:j] ………….. (i-1,j-k) ………….. ………….. i-1 i (i,j)
Corner Cutting ~100-1000 Better Numerical Search ~10-100 Ex.: good start guess, 28 evaluations, 3 iterations Accelleration of Pairwise Algorithm (From Hein,Wiuf,Knudsen,Moeller & Wiebling 2000) Simpler Recursion ~3-10 Faster Computers ~250 1991-->2000 ~106
a-globin (141) and b-globin (146) (From Hein,Wiuf,Knudsen,Moeller & Wiebling 2000) 430.108 : -log(a-globin) 327.320 : -log(a-globin -->b-globin) 747.428 : -log(a-globin, b-globin) = -log(l(sumalign)) l*t: 0.0371805 +/- 0.0135899 m*t: 0.0374396 +/- 0.0136846 s*t: 0.91701 +/- 0.119556 E(Length) E(Insertions,Deletions) E(Substitutions) 143.499 5.37255 131.59 Maximum contributing alignment: V-LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS--H---GSAQVKGHGKKVADALT VHLTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFS NAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR DGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH Ratio l(maxalign)/l(sumalign) = 0.00565064
VLSPADNAL.....DLHAHKR 141 AA long *########### …. ### 141 AA long 2 108 years 2 107 years 2 109 years *########### …. ### *########### …. ### ???????????????????? k AA long 109 years The invasion of the immortal link
Algorithm for alignment on star tree (O(length6))(Steel & Hein, 2001) *ACGC *TT GT s2 s1 a s3 *ACG GT *###### * (l/m)
Binary Tree Problem a1a2 * * # # # - - # # # - # TGA ACCT s1 s3 a1 a2 s2 s4 GTT ACG • The ancestral sequences & their alignment was known. ii. The alignment of ancestral alignment columns to leaf sequences was known The problem would be simpler if: How to sum over all possible ancestral sequences and their alignments?: A Markov chain generating ancestral alignments can solve the problem!!
- # # E # # - E * * lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) # # lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) _ #lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) # - lb Generating Ancestral Alignments a1 * a2 * # # l/m (1- lb)e-m E E (1- l/m) (1- lb) - # lb
The Basic Recursion ”Remove 1st step” - recursion: S E ”Remove last step” - recursion: Last/First step removal are inequivalent, but have the same complexities. First step algorithm is the simplest.
Sequence Recursion: First Step Removal Pa(Sk): Epifixes (S[k+1:l]) starting in given MC starts in a. Pa(Sk) = Where P’(kS i,H) = F(kSi,H)
Maximum likelihood phylogeny and alignment Gerton Lunter Istvan Miklos Alexei Drummond Yun Song Human alpha hemoglobin;Human beta hemoglobin; Human myoglobin Bean leghemoglobin Probability of data e-1560.138 Probability of data and alignment e-1593.223 Probability of alignment given data 4.279 * 10-15 = e-33.085 Ratio of insertion-deletions to substitutions: 0.0334
Metropolis-Hastings Statistical Alignment Lunter, Drummond, Miklos, Jensen & Hein, 2005