Human Genome Lecture
330 likes | 1.03k Vues
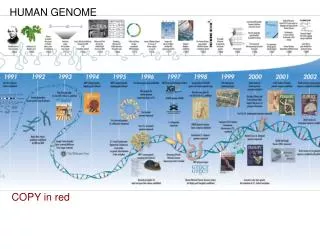
Human Genome Lecture. Historical aspects of the HGP EST sequencing: Finding new genes faster than ever Using 3’ ESTs to generate human gene maps First comprehensive genome-wide human gene maps Sequence of human genome Complex genomic regions and sequence limitations.

Human Genome Lecture
E N D
Presentation Transcript
Human Genome Lecture • Historical aspects of the HGP • EST sequencing: • Finding new genes faster than ever • Using 3’ ESTs to generate human gene maps • First comprehensive genome-wide human gene maps • Sequence of human genome • Complex genomic regions and sequence limitations
Key pre-HGP scientific advances • Structure of DNA determined (1953) • Watson & Crick • Recombinant DNA created (1972) • P. Berg; Cohen and Boyer • Methods for DNA sequencing developed (1977) • Maxam & Gilbert; F. Sanger • PCR invented (1985) • K. Mullis • Automated DNA sequencer developed (1986) • L. Hood
Obstacles to formation of the HGP 1) Financial/political: “Big biology is bad biology” -departure from cottage industry culture of biology -devoid of “hypothesis-driven” research -what will it cost? -will it take away $$ from other programs? 2) Why sequence the “Junk”? -protein coding regions make up <1.5% of the genome -waste of time/money to sequence repetitive, hard-to-sequence regions 3) It is impossible to do -mid 1980s: -primitive sequencing capabilities (500 bp/day/lab) -primitive computer capabilities/bioinformatics resources
Significance of the HGP • “The book of life”, “The grail of human biology”, “Code of codes” • The instructions to create a human being • The genome is a product of evolution - molecular replicator (DNA) + heritable variation + time + changing environment = genome - record of the evolutionary history of our species • Comparative genomics & “the genes that make us human” • The genome: unparalled system of information storage - 70 trillion cells in human body - each cell stores 3 billion units of information
Significance of the HGP (cont) • Biology in the 21st century: - equivalent of learning to read a new language • The genome as dynamic not static: - perspective on past/future of the species • Implications for health and disease: -Genetic disease gene discovery: single-gene diseases & multifactorial diseases -DNA-based diagnostics -New drug targets -Gene therapy implications -Therapeutic uses vs. enhancements • Accumulation of a molecular “parts lists” of human physiology & anatomy: - Lander: “Periodic Table of the Elements” analogy
Rapid Gene Identification & Mapping: ESTs and Gene-based STSs • Single-pass sequencing of randomly selected cDNA clones • Obtain sequences from 5’ and 3’ ends of cDNA inserts • Rapidly & cheaply identify human genes • Alzheimer’s gene discovered by EST database search • 3’UT sequence: ideal for STS development & PCR-based gene mapping • Readily scaled up for development of most comprehensive human gene maps (Science 1996, 1998)
“One gene = one STS” • Gene-based STSs as the basis for a human gene map • Berry et al, Nature Genetics 1995 • ESTablishing a human transcript map • Boguski and Schuler, Nature Genetics 1995
Size and gene content of the 24 human chromosomes. A, Size of each human chromosome, in millions of base pairs (1 million base pairs = 1 Mb). Chromosomes are ordered left to right by size. B, Number of genes identified on each human chromosome. Chromosomes are ordered left to right by gene content. (Based on www.ensembl.org, v36.)
Genomic sequencing vs EST sequencing • EST (single pass cDNA) sequencing - very fast but not error-free (e.g. 99% accuracy) - very rapid gene identification (reliance on mRNA) - cDNA abundance influences coverage some genes will be missed normalized cDNA libraries improve coverage provides a gene expression profile • Genomic sequencing - pre-2001: much slower method for gene finding -must do gene id by computer prediction - will generate complete gene and genome information, e.g. introns, regulatory regions, intergenic regions, repeats, etc. - more expensive way to id genes - independent of gene expression level concerns - highly accurate when complete
Table 11. Genome overview. Size of the genome (including gaps) 2.91 Gbp Size of the genome (excluding gaps) 2.66 Gbp Longest contig 1.99 Mbp Longest scaffold 14.4 Mbp Percent of A+T in the genome 54 Percent of G+C in the genome 38 Percent of undetermined bases in the genome 9 Most GC-rich 50 kb Chr. 2 (66%) Least GC-rich 50 kb Chr. X (25%) Percent of genome classified as repeats 35 Number of annotated genes 26,383 Percent of annotated genes with unknown function 42 Number of genes (hypothetical and annotated) 39,114 Percent of hypothetical and annotated genes with unknown function 59 Gene with the most exons Titin (234 exons) Average gene size 27 kbp Most gene-rich chromosome Chr. 19 (23 genes/Mb) Least gene-rich chromosomes Chr. 13 (5 genes/Mb), Chr. Y (5 genes/Mb) Total size of gene deserts (>500 kb with no annotated genes) 605 Mbp Percent of base pairs spanned by genes 25.5 to 37.8* Percent of base pairs spanned by exons 1.1 to 1.4* Percent of base pairs spanned by introns 24.4 to 36.4* Percent of base pairs in intergenic DNA 74.5 to 63.6* Chromosome with highest proportion of DNA in annotated exons Chr. 19 (9.33) Chromosome with lowest proportion of DNA in annotated exons Chr. Y (0.36) Longest intergenic region (between annotated + hypothetical genes) Chr. 13 (3,038,416 bp) Rate of SNP variation 1/1250 bp * In these ranges, the percentages correspond to the annotated gene set (26, 383 genes) and the hypothetical + annotated gene set (39,114 genes), respectively.
Significant findings arising from analysis of the draft sequence of the human genome • The genomic landscape shows marked variation in the distribution of a number of features, including genes, transposable elements, GC content, CpG islands and recombination rate. This gives us important clues about function. For example, the developmentally important HOX gene clusters are the most repeat-poor regions of the human genome, probably reflecting the very complex coordinate regulation of the genes in the clusters. • There appear to be about 30,000–40,000 protein-coding genes in the human genome—only about twice as many as in worm or fly. However, the genes are more complex, with more alternative splicing generating a larger number of protein products. • The full set of proteins (the 'proteome') encoded by the human genome is more complex than those of invertebrates. This is due in part to the presence of vertebrate-specific protein domains and motifs (an estimated 7% of the total), but more to the fact that vertebrates appear to have arranged pre-existing components into a richer collection of domain architectures. • Hundreds of human genes appear likely to have resulted from horizontal transfer from bacteria at some point in the vertebrate lineage. Dozens of genes appear to have been derived from transposable elements. • Although about half of the human genome derives from transposable elements, there has been a marked decline in the overall activity of such elements in the hominid lineage. DNA transposons appear to have become completely inactive and long-terminal repeat (LTR) retroposons may also have done so. • The pericentromeric and subtelomeric regions of chromosomes are filled with large recent segmental duplications of sequence from elsewhere in the genome. Segmental duplication is much more frequent in humans than in yeast, fly or worm. • Analysis of the organization of Alu elements explains the longstanding mystery of their surprising genomic distribution, and suggests that there may be strong selection in favour of preferential retention of Alu elements in GC-rich regions and that these 'selfish' elements may benefit their human hosts. • The mutation rate is about twice as high in male as in female meiosis, showing that most mutation occurs in males. • Cytogenetic analysis of the sequenced clones confirms suggestions that large GC-poor regions are strongly correlated with 'dark G-bands' in karyotypes. • Recombination rates tend to be much higher in distal regions (around 20 megabases (Mb)) of chromosomes and on shorter chromosome arms in general, in a pattern that promotes the occurrence of at least one crossover per chromosome arm in each meiosis. • More than 1.4 million single nucleotide polymorphisms (SNPs) in the human genome have been identified. This collection should allow the initiation of genome-wide linkage disequilibrium mapping of the genes in the human population.
Patterns of intrachromosomal and interchromosomal duplication in the human genome Bailey, et al, Science, 2002
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 0 50 100 base position (Mb) 150 200 250 HapMap phase 1 Distribution of >50 kb gaps in HapMap phase 1 - CEU chromosome lengths >50 kb gap between SNPs excluding centromere gaps heterochromatin T. Hudson
Genome “Structural Variation” • Broadest sense: all changes in the genome not due to single base-pair substitutions: • Copy number variations (CNVs) • CNV loci may cover 12% of genome • Insertions/Deletions (indels) • e.g. Repeats: STRs, VNTRs • Inversions • Duplications and translocations
Limitations of Genome Sequencing • Nexgen sequencers are short read • Repeated/duplicated sequences often can’t be positioned • Segmental duplications make up 5% of genome • >95% identity; >20kb • Smaller-size, highly duplicated sequence families exist • Complex, duplication-rich regions • >200 gaps (>50kb each) in human genome • Difficult to accurately assemble • Linked to many human diseases • Linked to evolutionary adaptation • Location of “missing heritability” of GWAS? • Are critical regions of the genome being missed/ignored?
Limitations of next-generation genome sequence assembly Can Alkan, Saba Sajjadian & Evan E Eichler Nature Methods Volume: 8, Pages: 61–65 Year published: (2011) DOI: doi:10.1038/nmeth.1527 Published online 21 November 2010 Abstract Abstract High-throughput sequencing technologies promise to transform the fields of genetics and comparative biology by delivering tens of thousands of genomes in the near future. Although it is feasible to construct de novo genome assemblies in a few months, there has been relatively little attention to what is lost by sole application of short sequence reads. We compared the recent de novo assemblies using the short oligonucleotide analysis package (SOAP), generated from the genomes of a Han Chinese individual and a Yoruban individual, to experimentally validated genomic features. We found that de novo assemblies were 16.2% shorter than the reference genome and that 420.2 megabase pairs of common repeats and 99.1% of validated duplicated sequences were missing from the genome. Consequently, over 2,377 coding exons were completely missing. We conclude that high-quality sequencing approaches must be considered in conjunction with high-throughput sequencing for comparative genomics analyses and studies of genome evolution.