Download

1 / 33
470 likes | 1.29k Vues
TUMS. Metabolism of Amino Acid (Carbon Skeletons) Part 2. Dr. Azin Nowrouzi Tehran University of Medical Sciences. Fate of the C-Skeleton of Amino Acids. 2. Outline of catabolism of 20 amino acids. 4. 5. Degradation of Carbon Skeletons. 6.

E N D
TUMS Metabolism of Amino Acid (Carbon Skeletons) Part 2 Dr. Azin Nowrouzi Tehran University of Medical Sciences
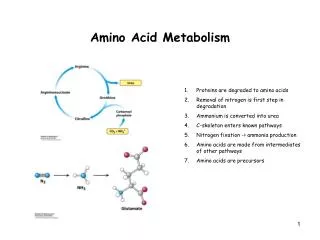
Degradation of Carbon Skeletons 6 • Seven products result from the catabolism of amino acid carbon skeletons: • oxaloacetate, α-ketoglutarate, pyruvate, fumarate, acetyl coA, acetoacetyl coA, succinyl coA • Glycogenic • Their catabolism produces pyruvate or one of the intermediates of the Crebs cycle. • These are substrates for gluconeogenesis • So they can produce glycogen in liver and muscle. • Lipogenic (or ketogenic) • Their catabolism produces acetoacetate or its precursors acetyl coA or acetoacetyl coA
Phenylketonuria (PKU) Disease • Deficiency of Phe hydroxylase • Occurs in 1:20,000 live births in U.S. • Seizures, mental retardation, brain damage • Treatment: limit phenylalanine intake • Screening of all newborns mandated in all states 11
Transfer of nitrogen components from tissues to the liver for urea synthesis
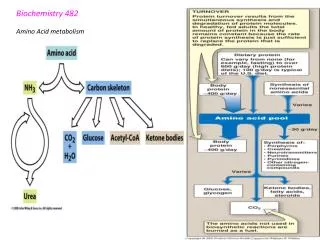
Fed state 16
Fasting (starvation) (i) For the first 7 days, maintain blood glucose (brain use 65% of glucose 400 - 600 Cal) (ii) > 7 days: Protein proteolysis decreases (protect essential proteins) therefore use over a prolonged period compromises organism. (iii) → Switch to Ketone bodies 17
AA are released from muscle during the post- absorptive state (O/N fast). Of the AA released by muscle Ala= 30% & Gln= 25% (total> 50%) But output (Ala+Gln) > abundance in muscle proteins which contain 7-10% Ala & 6% Gln Where does this Ala & Gln come from? 19
Sources of Alanine (from Muscle) (i)Muscle: Protein → Ala + aa aa→ NH4+ + α keto acids α keto acids → Ala (“simplest” aa). Therefore total Ala released > Ala derived from proteins (ii) Liver: Ala →NH4+ + α keto acids NH4+→ urea (iii) As well Glucose → Pyruvate (no N) → Ala (with N) Therefore Ala serves as a vehicle for transport of NH4+ from muscle to liver (NH4+ is generated through breakdown of aa →energy). (iv) Because free NH4+ is very toxic even at low levels therefore Pyruvate + NH4+ →Ala (non-toxic) (v) In liver: NH4+ → urea for excretion 20
Specialized Amino Acid Roles 1. Certain NEAA continue being synthesized even when adequate levels are supplied in diet because of a specialized role 2. ARG → urea synthesis ASP → urea synthesis GLU → conduit for disposal of N 3. ALA & GLN → key role in exchange between tissues (liver & skeletal muscle) 4. Liver: major site gluconeogenesis (AA → Glucose) major site urea synthesis (kidneys to a lesser extent) 5. Skeletal Muscle: 60% total body protein, 50% total body AA pool and is the major source to provide AA carbons → hepatic gluconeogenesis 21
Amino Acid Degradation 22 • Removal of alpha-amino groups • Nitrogen excretion • Fate of carbon skeletons
Removal of alpha-amino groups 23 • Mechanisms of –NH2 removal • Transamination • Oxidative deamination • Amino acid oxidases • Threonine or Serine dehydratase
A. Transamination Removal of Nitrogen by aminotransferase 24
L- and D- Amino Acid Oxidases • They are present in liver and kidneys. • They have low activity • Their physiologic value is not clear. Amino Acid + H2O α-ketoacid + NH3
D. Amino acid Dehydratase Threonine Urea 27 Serine and Threonine can be Directly Deaminated
Fate of Nitrogen in Different Organisms • Other excretion products • creatinine • uric acid 28
Disposal of amino group 29 • Urea cycle (Krebs-Henseleit cycle) • Provides 25-30 g of urea daily for urine formation in the kidneys • Carbamoyl Phosphate Synthetase • Ornitine Carbamoyl Transferase • Argininosuccinate Synthetase • Argininosuccinate Lyase • Arginase • Excretion of free ammonia • Glutamine synthetase • Glutaminase
Ammonium Ion is Converted into Urea Urea cycle 31