Download

1 / 56
570 likes | 722 Vues
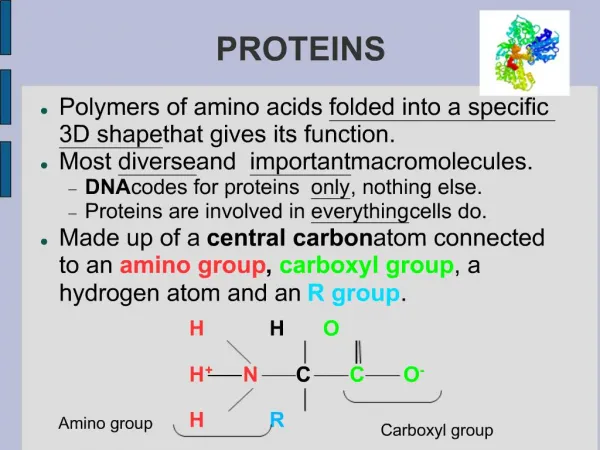
Proteins. Lecture 10. ביו-אינפורמטיקה של חלבונים Proteomics. פרוטאומיקה היא חקר התכונות של מגוון החלבונים המיוצרים ע"י אורגניזם בגישה שיטתית ומערכתית ועבודה ב High throughput מהם החלבונים שהאורגניזם מייצר? Gene prediction מהם המודיפיקציות שנעשות על החלבון?

E N D
Proteins Lecture 10
ביו-אינפורמטיקה של חלבונים Proteomics • פרוטאומיקה היא חקר התכונות של מגוון החלבונים המיוצרים ע"י אורגניזם • בגישה שיטתית ומערכתית ועבודה ב High throughput • מהם החלבונים שהאורגניזם מייצר? Gene prediction • מהם המודיפיקציות שנעשות על החלבון? • מהו המבנה השניוני של חלבונים אלו? • מהו המבנה המרחבי של חלבונים אלו? Structural genomics • מהו התפקיד של חלבונים אלו? Functional genomics • מהי תבנית הביטוי של חלבונים אלו?: Expression pattern • מהו מנגנון פעולתם? למשל אינטראקציות בין חלבונים. • איך החלבונים עוברים מודיפיקציה (פוספורילציה, גליקוליזציה וכו') • איך החלבונים מתפרקים וממוחזרים?
X-ray crystallography • Obtain an ordered protein crystal. • Check x-ray diffraction. • Analyze diffraction pattern and produce an electron density map. • Thread the known protein sequence into the density map. Tyrosine
NMR • Nuclear Magnetic Resonance measures the radio frequency absorption of different nuclei in the protein. • Measure the distances between different atoms in the protein. • Use thousands of distance measurements to construct a single protein model under the constraints. • X-ray crystallography is the most widely used method. • NMR is typically applicable to small proteins • Quaternary structure of large proteins (ribosomes, virus particles, etc) can be determined by electron microscopes.
Structural bioinformaticsביואינפורמטיקה מבנית • התמחות בשיטות ממוחשבות לאנליזה של מבנה חלבונים וDNA. • יכולת "לנבא" אינפורמציה מבנית ובכך לחסוך בניסוים ארוכים ויקרים. • יכולת לתכנן מבנים מולקולרים.
Swiss-Prot file format entry
מה אפשר ללמוד מהרצף הראשוני ? -משקל מולקולרי -ערכי PI -אזורים הידרופוביים/הידרופיליים -איזורי טרנסממברנליים -המצאות מוטיבים www.expasy.ch/tools/
PDB (Protein Data Bank )www.rcsb.org • holds 3D models of biological molecules (protein, RNA,DNA). • Contains almost 26,000 different models. Many entries are redundant,actually between 3000-5000 unique proteins are included. • Models are arranged in files (accession number, xyz coordinates for each atom). • PDB is growing fast, but the number of NEW folds discovered is not growing fast
PDB model: A model defines the 3D positions of atoms in one or more molecules: A protein, a protein complex, protein and DNA, etc. The models also include the positions of ligand molecules, solvent molecules, metal ions, etc. Cadmium ion Example – 1D66 (4 chains) : GAL4 S. cerevisiae transcription factor. Homo dimmer DNA
Terminology • Secondary structure: Loop Beta strand • Motif – a group of packed secondary structures. • Domain – A fundamental unit of the tertiary structure. The domain can fold independently and perform its function independently. Domains are made of one or more motifs. Alpha helix Beta-Alpha-Beta motif
visualizations • View by chain View by amino acid group (hydrophobic) • View by structure We can also examine the active site in detail. • View by solvent accessible surface. • View by surface and electrostatic potential
Flexprot: pairwise alignment. Rigid alignment Flexible alignment Structural alignment • Structural alignment is considered more accurate since, during evolution, the structure is more conserved than the sequence. • Input: PDB files Output: Possible alignments • The results can be viewed with Protein explorer.
Default parameters • Structural alignments were used to find the default parameters for the matrices of sequence alignment (PAM, blosum). ? = Possible alignment (Sequence alignment Different parameters) “True” alignment (structural alignment) No Yes Try new set of parameters Default parameters
Relationships between MSA and structure • Aligning similar proteins reveals conserved residues that are important for function or structure (recall: IPNS example).
Find homologue protein sequences (psi-blast) Perform multiple sequence alignment (removing doubles) Construct an evolutionary tree Calculate mutation rate for each site ConSurf :Working process Input a protein with a known 3D structure (PDB id or file provided by the user) Present calculations by coloring the 3D structure http://consurf.tau.ac.il/
Human (M) S. cerevisiae (G) Human (M) S. cerevisiae (M) Chimp (M) S. pombe (G) Chimp (G) S. pombe (G) Consurf- Why is phylogeny important? Example:Which of the two sites is more conserved? Or - Which site evolves faster?
Protein structure classification Why classify proteins Number of solved structures grow rapidly Generate overview of structure types Detect similarities (evolutionary relationships) Build model of a protein based on proteinsfrom the same class Set up prediction benchmarks When are two structures similar? • RMS of 6 Ang. – not related. RMS of 3-6 Ang – related RMS less than 3 Ang – similar • Two structures are of the same fold if they have RMS < 3 Ang over 70% of their length • Use the RMS measure (root mean square) for superposition of corresponding residues
SCOP Manual classification (A Murzin) http://scop.mrc-lmb.cam.ac.uk/scop/ CATH Semi manual classification (C orengo) FSSP Automatic classification (L Holm) Classification schemes 5 classes
Major classes in scop Classes All alpha proteins Alpha and beta proteins (a/b) Alpha and beta proteins (a+b) Multi-domain proteins Membrane and cell surface proteins Small proteins All alpha: Hemoglobin (1bab) All beta: Immunoglobulin (8fab) Alpha/beta: (1hti) Alpha+beta: Lysozyme (1jsf)
Folds - Proteins which have the same (>~50%) secondary structure elements arranged the in the same order in the protein chain and in three dimensions are classified as having the same fold. Superfamilies - superfamily contains proteins which are thought to be evolutionarily related due to (*)Sequence, (*)Function, and (*)Special structural features. Relationships between members of a superfamily may not be readily recognizable from the sequence alone Families - Contains members whose relationship is readily recognizable from the sequence (>~25% sequence identity) Families are further subdivided into Proteins Proteins are divided into Species The same protein may be found in several species
Folds: how many? • Chothia (1992) – appr. 1,000 folds • Estimates vary from 1,000 – 10,000 • With 100,000 human proteins, ~20 genes per fold on average Chothia, C., Proteins. One thousand families for the molecular biologist. Nature, 1992. 357(6379): p. 543-4. Zhang, C. and C. DeLisi, Estimating the number of protein folds. J Mol Biol, 1998. 284(5): p. 1301-5. Distribution of protein seqs. among protein families
Finding domains in your protein Only a fraction of the domains have been experimentally characterized, . If you find such a domain within your protein, you may get information, such as active site, fold, protein modification, interactions with protein/DNA molecules, ligands Collection of domains Each collection has pros and cons, using different modeling and curators…. “curated”: The families are created semi-automatically based on expert knowledge, sequence similarity, other databases The major domain servers: enable looking simultaneously at a few domain collections. Interpro- search in Prosite, prints, ProDom and Pfam CD-search – search in Pfam, CDD. Pfscan – search in the most updated PROSITE.
Prositehttp://www.expasy.ch/prosite • Characterization of protein families by conserved motifs (called patterns) observed in a multiple sequence alignments of known homologues. • Each family is defined by a single motif. • Pattern is a method for describing a conserved sequence described using regular expressions. • Entries are divided into two files • Pattern/profile file: the pattern and all SwissProt matches. • Documentation file: Details of the characterized family, a description of the biological role of the chosen motif, references. Example: [AC]-x-v-x(4)-{ED}. [Ala or Cys]-any-val-any-any-any-any-any but Glu or Asp
Modeling domain - example 1 consensus multiple alignment pattern [AC]-A-[GC]-T-[TC]-[GC] profile
Modeling domains • What is a “good” model? • How do we measure the success of a pattern? Sensitivity =TP/true Specificity=TP/positives modeling is a compromise between sensitivity and specificity (we want many true positive and few false positives)
True SensitivityTP / True SpecificityTP / Predicted TP Predicted Modeling domain - example 2 genome Additional true domains AGACT AGTCT ACAGT ACTGT False domains ACTCT AGTGT ACACT AGAGT • Consensus representation: • AGTCT • 0 mismatches search: • 1 true positive • 3 true negatives • No false positive • high specificity (1), low sensitivity(1/4) • 1 mismatch search: • 2 true positive • 2 true negative • 2 false positives • Specificity=1/2, sensitivity=1/2 Training set AGACT AGTCT ACAGT ACTGT AGTCT
True SensitivityTP / True SpecificityTP / Predicted TP Predicted Modeling domain - example 2 genome Additional binding sites AGACT AGTCT ACAGT ACTGT Non binding sites ACTCT AGTGT ACACT AGAGT • PSSM: • High probability search: • 4 true positive • 0 true negatives • 4 false positive • specificity=1/2, high sensitivity! (1) Training set AGACT AGTCT ACAGT ACTGT AGTCT If the model is more detailed: higher sensitivity, lower specificity.
Blocks http://www.blocks.fhcrc.org • Blocks are MSA corresponding to the most highly conserved regions of proteins. • Families are taken from InterPro. • Creation of BLOCKS by automatically detecting the most highly conserved regions of each protein family • Blocks of 5-200 aa long alignments. • A family is characterized by a group of common blocks.
Prints http://www.bioinf.man.ac.uk/ddbrowser/PRINTS/ • Fingerprint are made up from several conserved motifs. Pfam http://www.sanger.ac.uk/Software/Pfam • Pfam contains MSAs and HMM-profiles of complete protein domains.
איך קובעים את המבנה השניוני כאשר המבנה התלת מימדי ידוע? DSSP is a database of secondary structure assignments (and much more) for all protein entries in the Protein Data Bank (PDB). DSSP is also the program that calculates DSSP entries from PDB entries. The DSSP code (www.sander.ebi.ac.uk/dssp/ ) H = alpha helix B = residue in isolated beta-bridge E = extended strand, participates in beta ladder G = 3-helix (3/10 helix) I = 5 helix (pi helix) T = hydrogen bonded turn S = bend Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Kabsch and Sander Biopolymers 22:2577-2637, 1983 המבנה השניוני אינו "נמדד" בניסוי. אמנם בדרך כלל קל לאתר מבנים שניוניים בתוךהמבנה התלת-מימדי אבל לא תמיד הקביעה היא חד-משמעית. הבעיה חמורה במיוחדבקצוות של המבנים.
איך מנבאים את המבנה השניוני כאשר המבנה התלת מימדי לא ידוע? psa, sscp, sosui , PHD, PRIDCT • הרעיון הבסיסי: מסתבר שלחומצות אמיניות שונות יש נטיה שונה להמצא בכל מבנה שניוני • למבנים שניוניים יש אורך טיפוסי, ולכן הניבוי צריך להיות קונסיסטנטי למשך קטע • מחזוריות של חומצות הידרופוביות / הידרופיליות • נוכחות של חומצות פולריות קטנות (A,S,T) ובעיקר G אופיינית ל TURNS • Proline : בעל שרשרת ראשית מיוחדת ולכן יוצר kink , לא מתאים למשל למרכז של מבני Helix, מאד מתאים לקצה ה N-terminal של Helices
http://www.embl-heidelberg.de/predictprotein/predictprotein.htmlhttp://www.embl-heidelberg.de/predictprotein/predictprotein.html Input: sequence Output: Secondary structure prediction, globular regions, coiled-coil regions, transmembrane helices, PROSITE motifs, bound cystein…
הדיוק בניבוי איך קובעים הצלחה בניבוי? המדד המקובל בדר"כ Q3: אחוז החומצות שנקבעו נכון יחסית לכלל הניבוי MWHSGAVTTYPNKLYTREADSGGYVSAVL SequenceTHHHHHTTTEEEETTTEEEEETTTEEEET Prediction TTHHHHHHTTEEETTTEEEETTHHHHHTT Real Assignment Q3 is 18/30 = 0.6 בשנים האחרונות נבדקת הצלחת הניבויים כחלק מתחרויות CASP הבודקת באופן אוביקטיבי את יכולות הניבוי של מבנים שניוניים ושל המבנה התלת מימדי. יכולת הניבוי של המבנה השניוני עומדת כיום על 75-78 %