Protein Classification
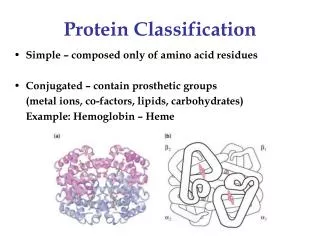
Protein Classification. Simple – composed only of amino acid residues Conjugated – contain prosthetic groups (metal ions, co-factors, lipids, carbohydrates) Example: Hemoglobin – Heme. Protein Classification. One polypeptide chain - monomeric protein More than one - multimeric protein

Protein Classification
E N D
Presentation Transcript
Protein Classification • Simple – composed only of amino acid residues • Conjugated – contain prosthetic groups (metal ions, co-factors, lipids, carbohydrates) Example: Hemoglobin – Heme
ProteinClassification • One polypeptide chain - monomeric protein • More than one - multimeric protein • Homomultimer - one kind of chain • Heteromultimer - two or more different chains (e.g. Hemoglobin is a heterotetramer. It has two alpha chains and two beta chains.)
Protein Classification Fibrous – • polypeptides arranged in long strands or sheets • water insoluble (lots of hydrophobic AA’s) • strong but flexible • Structural (keratin, collagen) Globular – • polypeptide chains folded into spherical or globular form • water soluble • contain several types of secondary structure • diverse functions (enzymes, regulatory proteins)
Protein Function • Catalysis – enzymes • Structural – keratin • Transport – hemoglobin • Trans-membrane transport – Na+/K+ ATPases • Toxins – rattle snake venom, ricin • Contractile function – actin, myosin • Hormones – insulin • Storage Proteins – seeds and eggs • Defensive proteins – antibodies
Globular Proteins Myoglobin/Hemoglobin Hemeproteins: group of specialized proteins that contain heme group as a tightly bound prosthetic group. prosthetic group: is a non-protein compound that is permanently associated with protein The role of heme group is dependent on the environment created by the three-dimensional structure of the protein. e.g. heme in cytochrome electron carrier, enzyme catalase active site Myoglobin and hemoglobin Oxygen carrier
Myoglobin/Hemoglobin • First protein structures determined • Oxygen carriers • Hemoglobin transport O2 from lungs to tissues • Myoglobin O2 storage protein
Structure and function of Hemoglobin Mb and Hb subunits structurally similar • 8 alpha-helices • Contain heme group • Mb monomeric protein • Hb heterotetramer (α22) - Hb found exclusively in red blood cells -transport O2 from lungs to capillaries of tissues and transfer CO2 from tissues to lungs -composed of 4 polypeptides held together by non-covalent interaction -each subunit is similar to myoglobin and contains a heme group hemoglobin myoglobin
-Myoglobin is O2 binding protein found in almost all mammals mainly in muscles and heart • -its main function is to store O2 for periods where energy demands is high, it also increases the rate of transport of oxygen within the muscle cells. • - Compact structure, 80% of its polypeptide chain is α-helix that labeled A to h that terminated by Proline or by -bends • The interior of the myoglobin is composed of NON-polar a.a. they packed together stabilized by hydrophobic interaction. • Charged a.a are located at the surface.
Structure of Heme Heme is a complex of protoporphyrine IX (4 pyrrole rings linked by methene bridges) and ferrous iron (Fe+2) Ferrous ion has 6 coordination bonds: 4 with the N of pyrrole rings and 2 are perpendicular one with N of histidine and the other is with O2 protoporphyrine IX porphyrine
Heme = Fe++ bound to tertapyrrole ring (protoporphyrin IX complex) Heme non-covalently bound to globin proteins through His residue O2 binds non-covalently to heme Fe++, stabilized through H-bonding with another His residue Heme group in hydrophobic crevice of globin protein
Heme group Distal histidine: stabilizes the binding of O2 to heme • Heme = Fe++ bound to tertapyrrole ring (protoporphyrin IX complex) • Heme non-covalently bound to globin proteins through His residue • O2 binds non-covalently to heme Fe++, stabilized through H-bonding with another His residue • Heme group in hydrophobic crevice of globin protein Proximal histidine
Distal histidine: stabilizes the binding ofO2 to heme Heme Oxygen Proximal histidine Ferrous ion Oxygen binding to Heme group
Hb tetramer can be described as two identical dimers, (α)1 and (α)2 The interaction between α and subunits is strong (hydrophobic, ionic and hydrogen interactions ) The interaction (α)1 and (α)2 is weak interaction (primary hydrophobic). The two dimers can move with respect to each other two conformations according to the presence or absence of O2
T-form (Taut or tense): the deoxy form of Hb. The two α dimers interact through a network of ionic bonds and hydrogen bonds that constrain the movement of the dimer. This form is the low O2 affinity form of Hb • R-form (relaxed form): the binding of O2 causes rupture of some ionic bonds and H-bonds the polypeptide chains have more freedom of movement. The R-form is the high O2 affinity form of Hb.
Oxygen binding to Myoglobin Degree of saturation Concentration of Oxygen (Partial pressure) Myglobin has one heme group bind only one oxygen molecule. Hemoglobin has 4 heme groups bind to 4 oxygen molecules O2 dissociation curve has hyperbolic shape Myglobin has higher O2 affinity than of Hemoglobin P50 of Mb is about 1 mmHg and for Hb is 26 P50 is O2 Partial pressure needed to half saturation of the Mb of Hb Oxygen dissociation curve
Oxygen transport proteins Efficient O2 transport protein should bind to O2 at high partial pressure (loading in lung) and release it (low affinity) at low Partial pressure of (unloading in the tissue)
Oxygen Binding Curves tissues • Mb has hyperbolic O2 binding curve • Mb binds O2 tightly. Releases at very low pO2 • Hb has sigmoidal O2 binding curve • Hb high affinity for O2 at high pO2 (lungs) • Hb low affinity for O2 at low pO2 (tissues) lungs Strong-binding Transition from weak to strong binding Weak-binding
O2 Binding to Hb shows positive cooperativity O2 Binding to Hb shows sigmoidal shape, low binding affinity at low con of Oxygen and high affinity at higher con cooperative binding by the four subunit of Hb The binding of one O2 molecule at one heme group increases the oxygen affinity of the remaining heme groups in the same hemoglobin molecule. The affinity of hemoglobin for the last O2 bound is 300 times greater than its affinity for the first O2 Hb O2 affinity increases as each O2 molecule binds Increased affinity due to conformation change Deoxygenated form = T (tense) form = low affinity Oxygenated form = R (relaxed) form = high affinity Increasing affinity for O2 Hemoglobin is efficient in delivering the O2 to the tissues from lung, myglobin which has hyperbolic O2-dissociation curve is unable to do that
Allosteric Interactions • Allosteric interaction occur when specific molecules bind a protein and modulates activity • Allosteric modulators or allosteric effectors bind reversibly to site separate from functional binding or active site • Modulation of activity occurs through change in protein conformation • 2,3 bisphosphoglycerate (BPG), CO2 and protons are allosteric effectors of Hb binding of O2
How is CO2 Exported? CO2 + H2O H2CO3 HCO3 + H+ Most of the CO2 produced in metabolism is hydrated and transported as bicarbonate ion. the hydration of CO2 by the zinc-dependent enzyme carbonic anhydrase.
+ O H C O H H O H H N C C P r o t e i n C N C C P r o t e i n 2 O R O R O A m i n o T e r m i n u s C a r b a m a t e o n A m i n o T e r m i n u s Binding of Hemoglobin to CO2 Some CO2 is carried as carbamate bound to the uncharged α-amino group Carbon dioxide is transported in the form of a carbamate on the amino terminal residues of each of the polypeptide subunits. Direct binding of CO2 to Hb stabilizes the T- form (deoxy) of Hb resulting in a decrease in its affinity for oxygen The formation of a carbamate also results in release of a proton into solution indirectly induces the Bohr effect
Bohr Effect • Increased CO2 leads to decreased pH CO2 + H2O <-> HCO3- + H+ • At decreased pH several key a.a’s protonated, causes Hb to be converted to T-conformation (low affinity) HbO2 + H HbH + O2. • Deoxy form of Hb has higher affinity of H than O2 • Protonation of some amino acids stabilizes the deoxyhemeglobin (T-form) • HCO3- combines with N-terminal α-amino group to form carbamate group. • Carbamation stabilizes T-conformation
Bisphosphoglycerate (BPG) • 2,3-Bisphosphoglycerate is an important allosteric effecter of hemoglobin Binding of BPG to Hb causes low O2 affinity • One molecule binds at the interface of all four subunits, and makes contacts with the β-subunits. BPG binds in the cavity between β-Hb subunits • Its binding stabilizes the deoxyhemoglobin state so stabilizes T-conformation. This promotes oxygen dissociation from oxyhemoglobin.
2,3-Bisphosphoglycerate • 2,3-BPG concentration increases in response to chronic hypoxia as in pulmonary obstruction or to high altitude or chronic anemia. • 2,3-BPG is present in erythrocytes at about 5 mM (at sea level), At high altitudes it is present at 8 mM. Shifts the curve to the right increase the O2 delivery to the tissue. • At high altitudes, wherein the partial pressure of oxygen is low, one would want hemoglobin to give up more of its bound oxygen to the tissues • Fetal Hb (α22) has low affinity for BPG, allows fetus to compete for O2 with mother’s Hb (α2 β2) in placenta • Role of 2,3-BPG in transfused blood
Carbon monoxide binding to Hb • CO binds tightly to one or more of heme iron forming carbon monoxyhemoglobin (HbCO) and hemoglobin is shifted to R-form causing the remaining heme with high O2 affinity shifts the O2-binding curve to the hyperbolic (left) inability of affected hemoglobin to deliver O2 to the tissue • Formation of methemoglobin: • oxidation of the heme component of Mb and Hb into ferric (Fe+3) state form metmyglobin and methmoglobin • The oxidized heme can't bind the O2 • This oxidation can result from drugs or toxins or from inherited defects • Occasional oxidation of heme is corrected by the enzyme NADH-cytochrome b5 reductase that found in the red blood cell. • Methemoglobin binds strongly to CN (poison that inhibits the cytochromal electron transport), so in the case of the cyanide poisoning amyle nitrite is taken which able to oxidize the heme group sequestering the CN
Types of Hemoglobins There are 4 different types of hemoglobins known: The most common is Hb A that form 90% of total Hb and consists of α2 β2 Hb F (α22) less than 2% Hb A2 (α22) 2-5% Hb A1C (α2 β2-glucose) 3-9 % Fetal hemoglobin (Hb F): tetramer α22 • Hb F is major Hb in the fetus and newborn. During the last month of pregnancy , it accounts for 60 % of the total Hb. • In the first few weeks of pregnancy embryonic Hb is synthesized Hb Gower1 (ζ2ε2) after that the liver starts HbF synthesis. After the development of the bone marrow the Hb A is synthesized at about the eighth month of the pregnancy and gradually replaces the Hb F Binding of the 2,3-BPG to HbF (α22) • HbF has higher affinity for O2 than dose HbA, bec it has lower binding affinity to 2,3-BPG and this facilitates the transfer of O2 from maternal circulation across the placenta to the red blood cells of the fetus • 2 globin chains (HbF) lack some positively charged amino acids found that found in the β globin (Hb A) reduce the 2,3BPG binding higher affinity to O2
Hemoglobin A2 (Hb A2 (α22)) • Hb A2 is a minor component of normal adult hemoglobin, appear firstly about 12 week after the birth and can form about 2% of the total Hb Hemoglobin A1c • Under physiologic conditions HbA is slowly and non-enzymatically glycosylated • The extent of glycosylation is dependent on the plasma level of particular hexoses • The most abundant glycosylated Hb is HbA1c which has glucose unit that covalently linked to amino group of N-terminal valines of the beta chain • In the case of Diabetes mellitus, the amount of HbA1c will increase
Hemoglobinopathies Defined as a family of disorders caused either by production of structurally abnormal hemoglobin molecule, synthesis of insufficient quantities of normal hemoglobin or rarely both • Sickle- cell anemia (HbS) • Hemoglobin C disease (HbC) • Thalassemia Sickle- cell anemia (Hemoglobin S disease “HbS”) • a glutamate residue is replaced by valine residue in the β-chains. This results in two fewer negative charges for the tetrameric structure. • The substitution of a hydrophobic amino acid for a hydrophilic one makes the resulting molecule “sticky.” This is because a hydrophobic patch has been created, which causes molecules to stick together at this point. This causes aggregation to occur in deoxyhemoglobin. • Subsequent to strand formation, several strands can assemble to form an insoluble fiber, which is what gives sickled cells there shape. • People with sickle cell anemia suffer from repeated crises brought on by physical exertion.
Hemoglobin C disease • HbC is a hemoglobin variant having a single substitution in the sixth position of the β-globin chain. In this case lysine is substituted. • Patients have a relatively mild chronic hemolytic anemia and they don't suffer from infractive crises Hemoglobin SC disease in this disease some β-globin chains have sickle-cell mutation and other β-globin chains carry mutation found in HbC Thalassemias • Thalassemia is a hereditary hemolytic disease in which an imbalance in the synthesis of globin chains occurs • Normally the synthesis of α-chains and β-chains are coordinated so that each α-globin has its β-globin • in the thalassemia the synthesis of either α- or β-globin chain is defective α-thalassemia: defect in the synthesis of the α-globin and there are 4 different levels of this type β-thalassemia:β-globin is decreased or absent, there are 2 different level of this type
2,3-bisphosphoglycerate Binding to Hemoglobin The negative charges on 2,3-bisphosphoglycerate interact with positive charges on hemoglobin (shown in blue) Shown here is the R state of hemoglobin, to which oxygen has a greater affinity. Notice how the binding site for BPG collapses. 2,3-bisphosphoglycerate binding to hemoglobin stabilizes the T state.
Sickle Cell Anemia • Sickle cell anemia was the first condition for which a genetic mutation was correlated with a physiological response. This is a homozygous recessive condition, in which offspring must inherit both of the mutated genes in order to develop the disease fully. • There are more than 300 different genetic variants of hemoglobin that are known. • In the case of sickle cell disease, a valine residue is substituted for a glutamate residue in the b chains. This results in two fewer negative charges for the tetrameric structure. • The substitution of a hydrophobic amino acid for a hydrophilic one makes the resulting molecule “sticky.” This is because a hydrophobic patch has been created, which causes molecules to stick together at this point. This causes aggregation to occur in deoxyhemoglobin. • Subsequent to strand formation, several strands can assemble to form an insoluble fiber, which is what gives sickled cells there shape. • People with sickle cell anemia suffer from repeated crises brought on by physical exertion. The hemoglobin content of their blood is about 1/2 of normal erythrocytes, and the sickled cells can block capillaries, causing severe pain.
Mutations in a- or b-globin genes can cause disease state • Sickle cell anemia – E6 to V6 • Causes V6 to bind to hydrophobic pocket in deoxy-Hb • Polymerizes to form long filaments • Cause sickling of cells • Sickle cell trait offers advantage against malaria • Fragile sickle cells can not support parasite
Structure and function of Hemoglobin -found exclusively in red blood cells -transport O2 from lungs to capillaries of tissues and transfer CO2 from tissues to lungs -composed of 4 polypeptides held together by non-covalent interaction -each subunit is similar to myoglobin and contains a heme group