Recombinant DNA Technology
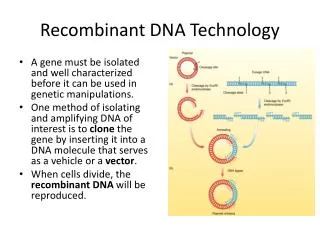
Recombinant DNA Technology. Dr. Hui LI Office : S408 Tel: 26538722. Process of cloning. Isolation of target gene Selection and construction of vectors Ligation of target DNA and vector Transformation of target gene into receptor cell S creening for recombinant plasmids


Recombinant DNA Technology
E N D
Presentation Transcript
Recombinant DNA Technology Dr. Hui LI Office : S408 Tel: 26538722
Process of cloning • Isolation of target gene • Selection and construction of vectors • Ligation of target DNA and vector • Transformation of target gene into receptor cell • Screening for recombinant plasmids • Expressing a cloned gene
§1. Isolation of target gene 1. Chemical synthesis only for simple polypeptide chain whose primary structure is clear. 2. Obtaining from genomic DNA library 3. Obtaining from cDNA library 4. polymerase chain reaction (PCR)
The genomic DNA library is a collection of the comprehensive DNA fragments representing the entire genome of a species.
mRNA Reverse transcripase The cDNA library represents the population of mRNAs, it only contains the exons of protein’s structural genes. cDNA replication dscDNA vector recombinate DNA E. coli recombinate DNA in E.coli
§1.1 Chemical synthesis of a gene • Oligonucleotide synthesis is the chemical synthesis of relatively short fragments of nucleic acids with defined chemical structure (sequence). • The technique is extremely useful in current laboratory practice because it provides a rapid and inexpensive access to custom-made oligonucleotides of the desired sequence. • Whereas enzymes synthesize DNA and RNA in a 5' to 3' direction, chemical oligonucleotide synthesis is carried out in the opposite, 3' to 5' direction. • Currently, the process is implemented as solid-phase synthesis using phosphoramidite method (固相亚磷酸三酯法). The occurrence of side reactions sets practical limits for the length of synthetic oligonucleotides (up to about 200 nucleotide residues) because the number of errors accumulates with the length of the oligonucleotide being synthesized. • Products are often isolated by HPLC to obtain the desired oligonucleotides in high purity.
Each nucleotide addition consists of four chemical reactions: deblocking (detritylation), coupling, oxidation, and capping. • During deblocking, DMT is removed from the growing oligonucleotide resulting in a free 5’ hydroxyl group. • During coupling, an activated nucleoside phosphoramidite is joined to the newly formed 5’ hydroxyl. • During oxidation, the unstable tricoordinated phosphate triester • linkage formed is converted to a protected tetracoordinated phosphate triester. • During capping, oligonucleotides that fail to join to an activated nucleoside are permanently blocked to prevent their further use in subsequent steps. • Figure adapted from (Caruthers et al. 1987). Oligonucleotide synthesis cycle
Oligonucleotide synthesis is carried out by a stepwise addition of nucleotide residues to the 5'-terminus of the growing chain until the desired sequence is assembled. Each addition is referred to as a synthetic cycle and consists of four chemical reactions: Step 1 - De-blocking (detritylation): The DMT group (二甲氧基三苯甲基)is removed with a solution of an acid, such as TCA (三氯乙酸),in an inert solvent and washed out, resulting in a free 5' hydroxyl group on the first base; Step 2 - Coupling: A nucleoside phosphoramidite (or a mixture of several phosphoramidites亚磷酰胺 ) is activated by an acidic azoles catalyst, tetrazole (四唑), 2-ethylthiotetrazole, 2-bezylthiotetrazole, 4,5-dicyanoimidazole, or a number of similar compounds. This mixture is brought in contact with the starting solid support (first coupling) or oligonucleotide precursor (following couplings) whose 5‘-hydroxy group reacts with the activated phosphoramidite moiety of the incoming nucleoside phosphoramidite to form a phosphite triester linkage(亚磷酸三酯键). This reaction is very rapid and requires, on small scale, about 20 s for its completion. The phosphoramidite coupling is also highly sensitive to the presence of water and is commonly carried out in anhydrous acetonitrile( 乙腈). Unbound reagents and by-products are removed by washing
Step 3 - Capping: After the completion of the coupling reaction, a small percentage of the solid support-bound 5'-OH groups (0.1 to 1%) remains unreacted and needs to be permanently blocked from further chain elongation to prevent the formation of oligonucleotides with an internal base deletion commonly referred to as (n-1) shortmers. This is done by acetylation (乙酰化作用)of the unreacted 5'-hydroxy groups using a mixture of acetic anhydride and 1-methylimidazole as a catalyst. Excess reagents are removed by washing. Step 4 - Oxidation: The newly formed tricoordinated phosphite triester linkage (亚磷酸三酯键)is not natural and is of limited stability under the conditions of oligonucleotide synthesis. The treatment of the support-bound material with iodine and water in the presence of a weak base (pyridine, lutidine, or collidine) oxidizes the phosphite triester into a tetracoordinated phosphate triester (磷酸三酯键), a protected precursor of the naturally occurring phosphate diester internucleosidic linkage.
Synthetic cycle for preparation of oligonucleotides by phosphoramidite method
(Left) Polymerase Cycling Assembly (PCA) synthesis procedure. Oligonucleotides with complementary regions are pooled and repeated annealing and polymerase extension cycles assembly and amplify a full-length product. (Right) Ligase Cycling Assembly (LCA) synthesis procedure. Using a thermostable ligase, repeated annealing and ligation cycles join oligonucleotides into increasingly larger strands. A final PCR step is often employed to amplify the full-length target from the incomplete products. Overview of PCA and LCA.
Normally, at the end of each procedure, 30-50 oligonucleotides are synthesized. They are most commonly used as antisense oligonucleotides, small interfering RNA, primers for DNA sequencing and amplification, probes for detecting complementary DNA or RNA via molecular hybridization, tools for the targeted introduction of mutations and restriction sites, and for the synthesis of artificial genes.
The polymerase chain reaction (PCR) is a rapid and versatile in vitro method for amplifying DNA. 1.2 Polymerase Chain Reaction
DNA template A pair of primers DNA polymerase (ex. Taq) dNTPs Mg2+-containing buffer PCR reaction system
Denaturing: the template DNA is denatured to become ssDNA from dsDNA by heating. Annealing: this step allows the hybridization of the primers with target DNA. Extension: this process is the DNA synthesis step. Procedures of PCR
§1.3 Isolating a gene from gene library Gene library:a collection of different DNA sequence from an organism, each of which has been cloned into a vector for ease of purification, storage and analysis. Genomic libraries (made from genomic DNA) Gene library cDNA libraries (made from cDNA- copy of mRNA)
§1.3.1Genomic DNA libraries Purify genomic DNA: prokaryotes or eukaryotes; Fragment this DNA : physical shearing and restriction enzyme digestion; Clone the fragments into vectors; Transfer the constructed vectors into recipient cells; Culture and amplify the clones; Screen the clone of target.
To make a representative genomic libraries , genomic DNA must be purified and then broken randomly into fragments that are correct in size for cloning into the chosen vector. Purification of genomic DNA : Eukaryotes:preparecell nuclei removeprotein, lipids and other unwanted macro- molecules by protease digestion and phase extraction. Prokaryotes:extracted DNA directly from cells
Genomic libraries Break DNA into fragments randomly: Physical shearing : pipeting, mixing or sonicaion Restriction enzyme digestion: partial digestion is preferred to get a greater lengths of DNA fragments.
Genomic libraries Selection of restriction enzyme • Ends produced (sticky or blunt) & • The cleaved ends of the vector to be cloned Sau3A: 5’-/GATC-3’, less selectivity BamH1: 5’-G/GATCC • Whether the enzyme is inhibited by DNA modifications (CpG methylation in mammals); • Time of digestion and ratio of restriction enzyme to DNA is dependent on the desired insert size range.
Genomic libraries Vectors According to genome’s size,we can select a proper vector to construct a library . Vectors Plasmid λ phage cosmid YAC insert (kb) 5 23 45 1000 The most commonly chosen genomic cloning vectors are λ replacement vectors which must be digested with restriction enzymes o produce the two λ end fragment or λ arms between which the genomic DNA will be digested
Genomic libraries λ phage vector in cloning Long (left) arm short (right) arm cos Exogenous DNA (~20-23 kb) cos short (right) arm Long (left) arm cos cos Exogenous DNA (~20-23 kb)
λ replacement vector cloning 0.preparation of arm and genomic inserts 2. Packingwith a mixture of the phage coat proteins and phage DNA-processing enzymes • Ligation 3.Infection and formation of plaques Library constructed
§1.3.2 cDNA libraries I2-1 mRNA isolation, purification I2-2 Check theRNA integrity I2-3 Fractionate and enrich mRNA I2-4 Synthesis of cDNA I2-5 Treatment of cDNA ends I2-6 Ligation to vector
cDNA libraries cDNA libraries • No cDNA library was made from prokaryotic mRNA. • Prokaryotic mRNA is very unstable • Genomic libraries of prokaryotes are easier to make and contain all the genome sequences.
cDNA libraries cDNA libraries • cDNA libraries are very useful for eukaryotic gene analysis • Condensed protein encoded gene libraries, have much less junk sequences. • cDNAs have no introns genes can be expressed in E. coli directly • Are very useful to identify new genes • Tissue or cell type specific (differential expression of genes)
cDNA libraries • Most eukaryotic mRNAs are polyadenylated at their 3’ ends 5’ cap AAAAAAAAAAn • oligo (dT) can be bound to the poly(A) tail and used to recover the mRNA. mRNA isolation
cDNA libraries Three methods to isolate mRNA. 1.Traditionally method was done by pass a preparation of total RNA down a column of oligo (dT)-cellulose 2.More rapid procedure is to add oligo(dT) linked to magnetic beads directly to a cell lysate and ‘pulling out’ the mRNA using a strong magnet 3.Alternative route of isolating mRNA is lysing cells and then preparing mRNA-ribosome complexes on sucrose gradients
cDNA libraries Check the mRNA integrity Make sure that the mRNA is not degraded. Methods: Translating the mRNA : use cell-free translation system as wheat germ extract or rabbit reticulocyte lysate to see if the mRNAs can be translated Analysis the mRNAs by gel elctrophoresis: use agarose or polyacrylamide gels
cDNA libraries Cloning the particular mRNAs Is useful especially one is trying to clone a particular gene rather to make a complete cDNA library. Fractionate on the gel:performed on the basis of size, mRNAs of the interested sizes are recovered from agarose gels Enrichment:carried out by hybridization Example: clone the hormone induced mRNAs (substrated cDNA library)
cDNA libraries Synthesis of cDNA : First stand synthesis:materials as reverse transcriptase ,primer( oligo(dT) or hexanucleotides) and dNTPs (Fig 1) Second strand synthesis:best way of making full-length cDNA is to employs a ribonuclease ( RNase H ) which recognises the RNA component of a DNA:RNA hybrid and cleaves the RNA at a number of non-specific sites .(Fig2)
cDNA libraries Treatment of cDNA ends Blunt end ligation of large fragment is not efficient, so we have to use special acid linkers to create sticky ends for cloning. The process : Move protruding 3’-ends(strand-special nuclease) Fill in missing 3’ nucleotide(klenow fragment of DNA polyI and 4 dNTPs) Ligate the blunt-end and linkers(T4 DNA ligase) Tailing with terminal transferase or using adaptor molecules Restriction enzyme digestion(E.coRI )
cDNA libraries Ligation to vector Any vectors with an E.coRI site would suitable for cloning the cDNA. The process : Dephosphorylate the vector with alkaline phosphatase Ligate vector and cDNA with T4 DNA ligase (plasmid or λ phage vector)
Screening procedures 1.2.3 Screening procedures 1 Screening 2 Colony and plaque hybridization 3 Expression screening 4 Hybrid arrest and release 5 Chromosome walking (repeat screening)
Screening procedures 1. Screening The process of identifying one particular clone containing the gene of interest from among the very large number of others in the gene library . • Using nucleic acid probe to screen the library based on hybridization with nucleic acids. • Analyze the protein product.
Screening procedures Screening libraries Searching the genes of interest in a DNA library • Hybridization to identify the interested DNA or its RNA product • Radiolabeled probes which is complementary to a region of the interested gene • Probes: • An oligonucleotide derived from the sequence of a protein product of the gene • A DNA fragment/oligo from a related gene of another species • Blotting the DNA or RNA on a membrane • Hybridize the labeled probe with DNAmembrane (Southern) or RNA (Northern) membrane
Screening procedures 2 Colony and plaque hybridization Transfer the DNA in the plaque or colony to a Nylon or nitrocellulose membrane Phage DNA bind to the membrane directly Bacterial colonies must be lysed to release DNA on the membrane surface. Hybridization (in a solution Containing Nucleic acid probe) (Alkali treatment) X-ray film(radio- actively labeled ) antibody or enzyme (modified nucleotide labeled Wash to remove unhybri- dization probe and visualize Line up the hybridizated region or repeated hybridization
Screening procedures Transfer to nitrocellulose or nylon membrane Keep master plate Select positive from master plate Denature DNA(NaOH) Bake onto membrane Probe with 32p-labled DNA complementary to gene of interest Expose to film Screening by plaque hybridization
Screening procedures 3 Expression screening • Identify the protein product of an interested gene • Protein activity • Western blotting using a specific antibody
Screening procedures Expression screening (1) If the inserts are cloned into an expression sites, it may be expressed. Therefore, we can screen for the expressed proteins. However, this screening may miss the right clone Example: the EcoRI site of lgt11 vector. The inserted genes have one in six change (1/6) to be in both the correct orientation (2 possibilities; ) and reading frame (three possibilities; three nucleotide code XXX).
Screening procedures Expression screening (2) Antibodies can be used to screen the expression library. The procedurehas similarities to the plaque hybridization protocol. ‘Plaque lift’ ( taken by placing a membrane on the dish of plaque) Immersedin a solution of the antibody Detectedby other antibodies Repeat cycles of screening to isolate pure plaques
Screening procedures 4 Hybrid arrest and screen Individual cDNA clones or pools of clones can be used to hybridize to mRNA preparation Hybrid arrest :translate the mRNA population directly, and the inhibition of translation of some products detected. Hybrid release translation : purify the hybrids and the hybridized mRNAs released from them and translated, it identifies the protein encoded by the cDNA clone
Screening procedures 5 Chromosome walking Definition: To clone the desired gene by repeated isolating adjacent genomic clones from the library. to obtain overlapping genomic clones that represent progressively longer parts of a particular chromosome .
Screening procedures Process: 1. Prepare a probe from the end insert . 2.The probe are used to re-screen the library by colony or plaque hybridization 3.Analyzed the new isolate clones and posited them relative to the starting clone. some will be overlapping. 4. Repeated the whole process using a probe from the distal end of the second clone.